Simpson Golabi Behmel syndrome
Simpson-Golabi-Behmel syndrome is a rare genetic condition that affects many parts of the body and occurs primarily in males. Simpson Golabi Behmel syndrome is classified as an overgrowth disorder, which means that affected infants are considerably larger than normal at birth (macrosomia) and continue to grow and gain weight at an unusual rate 1. The severity varies from very mild forms in carrier females to infantile lethal forms in affected males. The infantile lethal form of Simpson Golabi Behmel syndrome is sometimes known as Simpson Golabi Behmel syndrome type 2 2. The other signs and symptoms of Simpson-Golabi-Behmel syndrome vary widely. People with mild cases often live into adulthood.
People with Simpson-Golabi-Behmel syndrome have distinctive facial features including widely spaced eyes (ocular hypertelorism), an unusually large mouth (macrostomia), a large tongue (macroglossia) that may have a deep groove or furrow down the middle, a broad nose with an upturned tip, and abnormalities affecting the roof of the mouth (the palate). The facial features are often described as “coarse” in older children and adults with this condition.
Other features of Simpson-Golabi-Behmel syndrome involve the chest and abdomen. Affected infants may be born with one or more extra nipples, an abnormal opening in the muscle covering the abdomen (diastasis recti), a soft out-pouching around the belly-button (umbilical hernia), or a hole in the diaphragm (a diaphragmatic hernia) that allows the stomach and intestines to move into the chest and crowd the developing heart and lungs 3.
Simpson-Golabi-Behmel syndrome can also cause heart defects, malformed or abnormally large kidneys, an enlarged liver and spleen (hepatosplenomegaly), and skeletal abnormalities. Additionally, Simpson Golabi Behmel syndrome can affect the development of the gastrointestinal system, urinary system, and genitalia. Some people with Simpson Golabi Behmel syndrome have mild to severe intellectual disability, while others have normal intelligence.
About 10 percent of people with Simpson-Golabi-Behmel syndrome develop cancerous or noncancerous tumors in early childhood. The most common tumors are a rare form of kidney cancer called Wilms tumor and a cancerous tumor called a neuroblastoma that arises from developing nerve cells.
The incidence of Simpson-Golabi-Behmel syndrome is unknown. At least 250 people worldwide have been diagnosed with Simpson Golabi Behmel syndrome 1.
Simpson Golabi Behmel syndrome can be caused by mutations in the GPC3 and GPC4 genes. Mutations in other genes have been studied, but have in most instances have only been described in one person or one family. In other cases, the cause is unknown 1. Simpson Golabi Behmel syndrome is inherited in an X-linked manner 4.
Although there is no specific treatment or cure, there can be ways to manage the symptoms. A team of doctors is often needed to figure out the treatment options based on each person’s symptoms.
Figure 1. Simpson Golabi Behmel syndrome
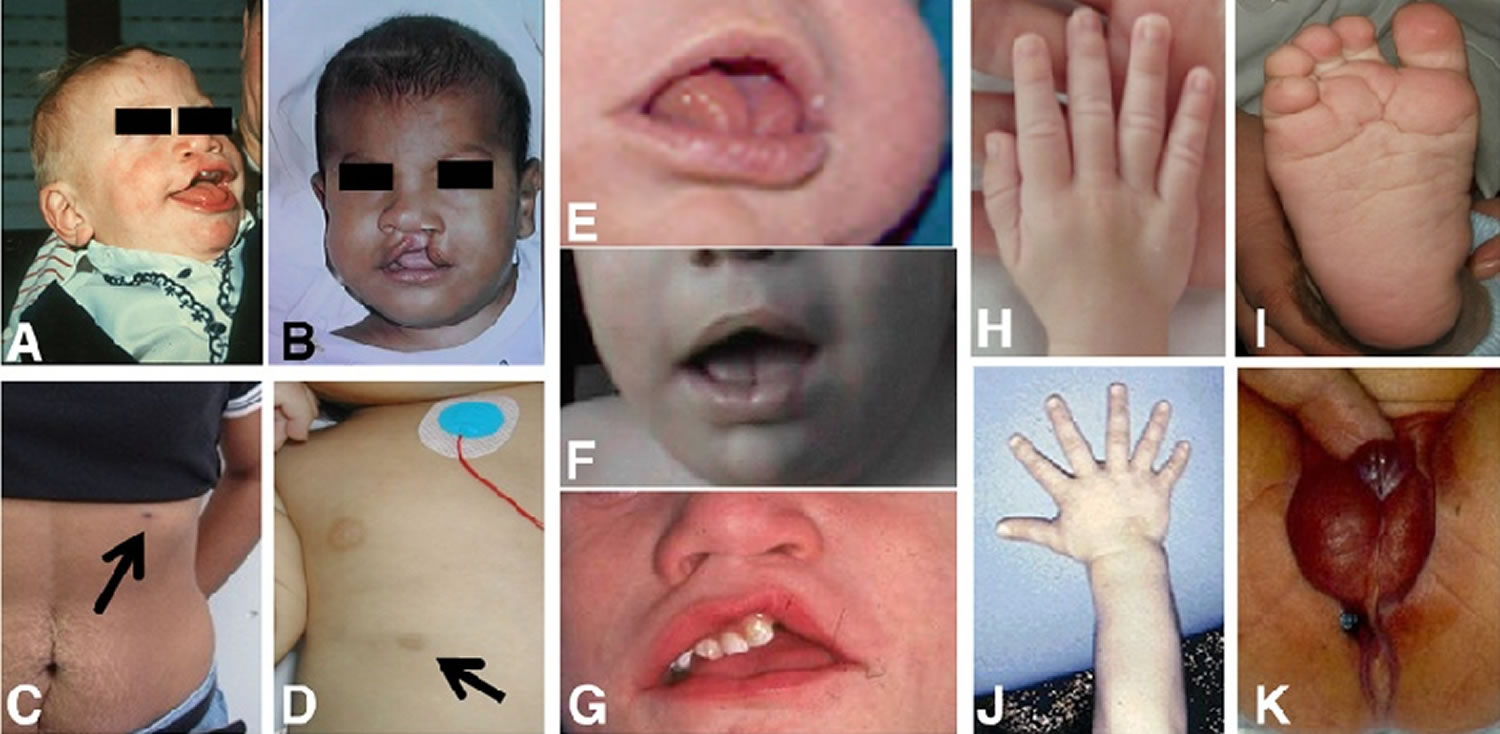
Footnote: Clinical findings in Simpson-Golabi-Behmel syndrome. A and B: Facial phenotype. Note the cleft lip, coarse, square face and broad nose. C and D: extra-nipple in a carrier mother and a toddler. E, F and G: close up of the mouth of three different patients. Note the large tongue, middle groove in the tongue, teeth malposition and repaired cleft (in G). H and J: Hands. Note broad hands and polydactyly in one subject. I: Deep plantar creases. K: abnormal genitalia in a male with hypospadias and proximal anal placement.
[Source 2 ]Simpson Golabi Behmel syndrome causes
Mutations in the glypican 3 (GPC3) gene are the most common cause of Simpson-Golabi-Behmel syndrome. The GPC3 gene provides instructions for making a protein called glypican 3, which blocks (inhibits) a developmental pathway called the hedgehog signaling pathway. This pathway is critical for cell growth and division (proliferation), cell specialization, and the normal shaping (patterning) of many parts of the body during embryonic development. Researchers believe that glypican 3 also helps establish the body’s shape by causing certain cells to self-destruct (undergo apoptosis) when they are no longer needed.
GPC3 gene mutations prevent glypican 3 from inhibiting the hedgehog signaling pathway. The resulting overactivity of this pathway leads to an increased rate of cell growth and division starting before birth. This increased cell proliferation accounts, at least in part, for the overgrowth that occurs in Simpson-Golabi-Behmel syndrome. It is unclear how changes in hedgehog signaling contribute to the other abnormalities that can occur with this disorder.
Some individuals with Simpson-Golabi-Behmel syndrome do not have an identified mutation in the GPC3 gene. Mutations in other genes have been studied as possible causes of this condition, but most of these genetic changes have been described only in single families or have not been confirmed in subsequent studies. In some cases, the cause of the condition is unknown.
Researchers have described a disorder with features overlapping those of Simpson-Golabi-Behmel syndrome, which they designated as Simpson-Golabi-Behmel syndrome type 2 (SGBS2). The signs and symptoms of this disorder are more severe than those that typically occur with Simpson-Golabi-Behmel syndrome, and affected individuals live only into infancy. Simpson Golabi Behmel syndrome type 2 is caused by mutations in the genes OFD1 and PIGA, also located on the X chromosome.
Simpson Golabi Behmel syndrome inheritance pattern
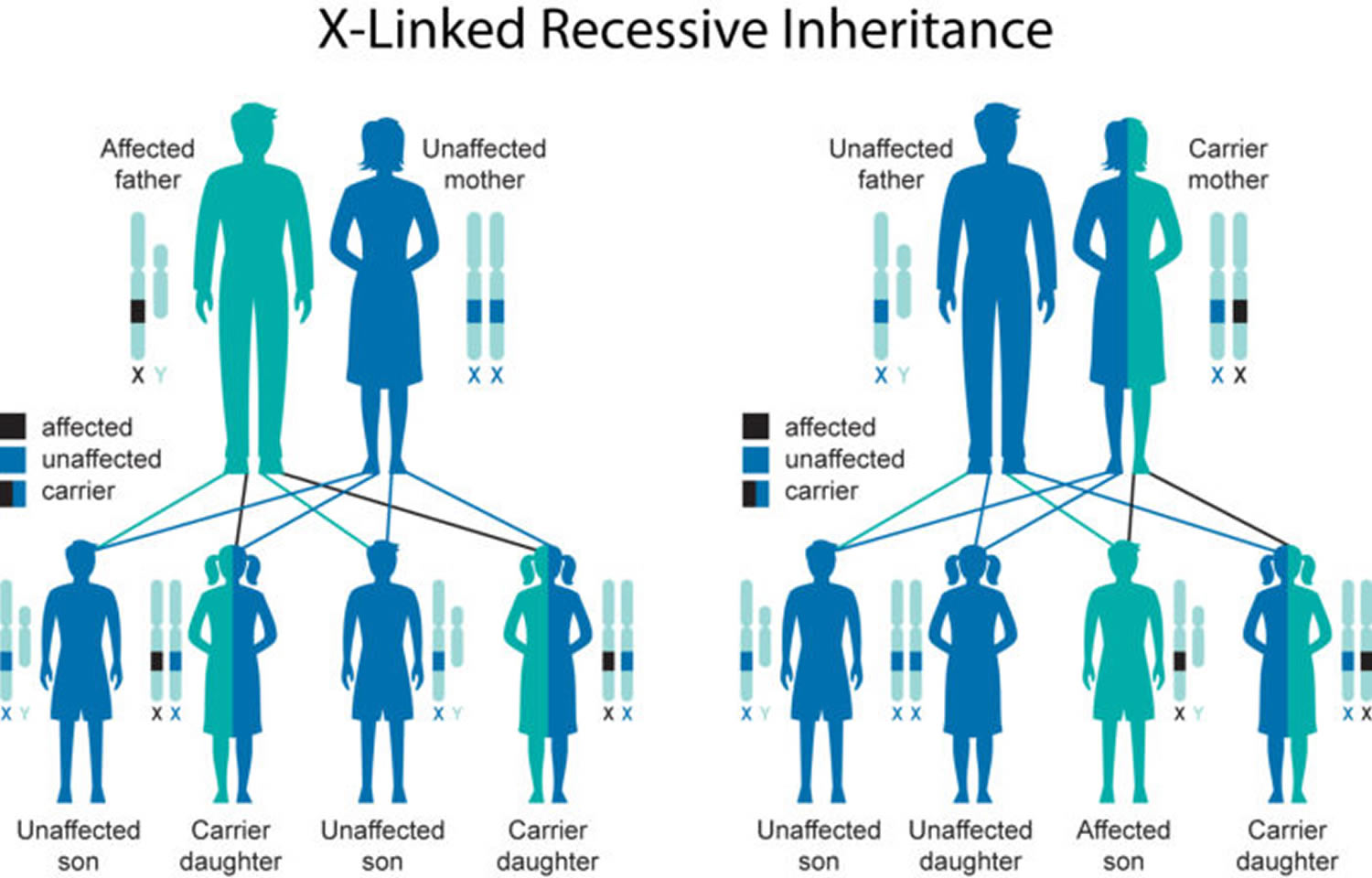
Simpson Golabi Behmel syndrome is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. Because females have two copies of the X chromosome, one altered copy of the gene in each cell usually leads to less severe health problems in females than in males, or it may cause no signs or symptoms at all.
Some females who have one altered copy of the GPC3 gene have distinctive facial features including an upturned nose, a wide mouth, and a prominent chin. Their fingernails may be malformed and they can have extra nipples. Skeletal abnormalities, including extra spinal bones (vertebrae), are also possible in affected females. Other females who carry one altered copy of the GPC3 gene do not have any health problems associated with Simpson-Golabi-Behmel syndrome.
Figure 2. Simpson Golabi Behmel syndrome X-linked inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Simpson Golabi Behmel syndrome signs and symptoms
Simpson-Golabi-Behmel syndrome type 1
Many different parts of the body can be affected when a person has Simpson Golabi Behmel syndrome. Not every person with Simpson Golabi Behmel syndrome has the same symptoms, and none have all of the symptoms listed here 5.
Overall
- General muscle weakness and low muscle tone (hypotonia) in 61% of people
- Large size (macrosomia)
Head
- Abnormal shape of the skull due to early bone fusion (craniosynostosis)
- Large jaw (macrognathia) in more than half of affected people
- Large sized head (macrocephaly) in the majority of affected people
- Specific facial feature that don’t look like other family members
- Large forehead
- Large and thick nose and lips
- Large tongue with a groove in the middle going from the front to the back in the majority of people
- Opening in the roof of the mouth (cleft palate) in 1 out of 4 people
- Split opening in the lip (cleft lip) in 1 out of 4 people
Central nervous system
- Attention deficit hyperactivity disorder (ADHD)
- Build-up of fluid in the brain (hydrocephalus)
- Disorder that causes breathing to pause or slow down during sleep (obstructive sleep apnea)
- Intellectual disability in about half of people
- Seizure disorder (epilepsy)
Speech and Language
- Speech disorder in the majority of people
Heart
- Any type of abnormality of the heart in about 1 out of 3 of people
Abdomen
- A weak area or defect in the belly that allows organs to push through (abdominal wall defect) in 1 out of 3 people
- Abnormal opening in the muscle between the chest and abdomen that helps with breathing and is present at birth (congenital diaphragmatic hernia) occurs rarely
- Extra nipples (supernumerary nipples) in half of people
- Kidney abnormalities in 33% of people
- Large kidney (nephromegaly) in half of people
- Large liver (hepatomegaly) in half of people
- Large spleen (splenomegaly)
- Organs that are larger than they should be (organomegaly) in most people
Genitals
- Opening of where the urine comes out (urethra) on the underside of the penis instead of at the tip (hypospadias)
- Testes that do not descend properly and are still inside the body (cryptorchidism)
Skeletal
- Abnormalities of the hands and feet
- Broad thumbs
- Extra fingers or toes (polydactyly)
- Large hands
- Short fingers and toes (brachydactyly) in half of people
- Underdevelopment of the pointer finger (index finger hypoplasia)
- Webbed or conjoined 2nd and 3rd fingers (syndactyly of the 2nd-3rd fingers)
- Chest deformity in less than half of people
- Rib malformations
- Scoliosis in one out of ten people
Tumors
- A 10% risk for certain tumors including:
- Adrenal neuroblastoma (a cancer found in the adrenal glands on top of the kidneys)
- Gonadoblastoma (a tumor with cells from the early testes or ovaries)
- Hepatoblastoma (cancerous liver tumor)
- Hepatocellular carcinoma (most common form of liver cancer)
- Medulloblastoma (a brain cancer that starts in the brain at the base of the skull)
- Wilms tumor (a kidney cancer seen in children)
Pregnancy
- Birth before 37 weeks (prematurity) in half of people
- Low blood sugar as a newborn (neonatal hypoglycemia) in 1 in 4 people
- More amniotic fluid than there should be (polyhydramnios) in the majority of people
Simpson-Golabi-Behmel syndrome type 2
Simpson Golabi Behmel syndrome type 2 is more serious and rarer than type 1. Boys with Simpson Golabi Behmel syndrome type 2 usually die a few months after birth. Most affected boys are born with extra fluid in multiple parts of the body (hydrops fetalis) and they also can have problems with their bones, distinct facial features, issues with organs and other medical problems.
Simpson Golabi Behmel syndrome diagnosis
Diagnosis of Simpson Golabi Behmel syndrome type 1 is made based on physical features of the patient, family history and genetic testing. There are no official criteria that are used to diagnose the condition. The main physical features are the different types of overgrowth (large body, large head, large fetus, large baby), the specific facial features, abnormalities that happen in the middle of the body (midline defects), and risk for tumors. The other physical features that are considered are organs larger than expected (organomegaly), issues with the skeleton, and problems with the heart, central nervous system, kidney, and gastrointestinal tract that are present from birth. A family history showing an X-linked pattern of inheritance can help with diagnosis. Genetic testing can involve sequencing and deletion/duplication analysis of the GPC3 gene, a chromosomal microarray, or a multigene panel that includes GPC3, GPC4, and other genes related to differential diagnoses.
Table 1. Recommended evaluations following initial diagnosis in individuals with Simpson-Golabi-Behmel syndrome type 1
| System/Concern | Evaluation | Comment |
|---|---|---|
| Oropharynx | Assess for macroglossia & orofacial clefting. | Referral to craniofacial team, incl feeding specialists |
| Eyes | Ophthalmologic examination | |
| Ears/Hearing | Audiologic evaluation | |
| Cardiac | Consider chest radiograph, EKG, & echocardiogram. | To evaluate for structural heart defects & conduction abnormalities |
| Respiratory | Assess for upper-airway sufficiency & signs/symptoms of sleep apnea; formal sleep study should be considered. | Particularly in those w/hypotonia & macroglossia |
| Renal | Examination for hypospadias & undescended testes in males | Referral to urologist, as needed |
| Renal ultrasound to assess for renal anomalies | ||
| Abdomen/Pelvis | Abdominal/pelvic ultrasound to initiate tumor screening | Further studies (e.g., MRI) may be indicated if findings are suspicious for a tumor. |
| Measurement of serum alpha fetoprotein | As a baseline screen for hepatoblastoma | |
| Musculoskeletal | Clinical evaluation for scoliosis | Particularly during times of rapid growth |
| Neurologic | Neurologic evaluation, head MRI, &/or EEG | If concerns for seizures |
| Endocrinologic | Assessment for hypoglycemia | In neonates |
| Miscellaneous/ Other | Developmental assessment | Incl speech & language assessment |
| Consultation w/clinical geneticist &/or genetic counselor |
Simpson Golabi Behmel syndrome treatment
The treatment for Simpson Golabi Behmel syndrome is based on the type of symptoms that each individual patient has. In the newborn period, right after birth, the baby’s blood sugar levels should be monitored to make sure they are not too low (hypoglycemia). The baby should also be checked to see if they are having problems breathing (airway obstruction). If the baby has any problems with their kidneys (renal anomalies) then their kidney function should also be monitored. Physical examinations should be done to check for a sideways curvature of the spine (scoliosis) during periods of time when the child is growing quickly. Social and intellectual development should also be monitored routinely.
Individuals with cleft palate require the coordinated efforts of a team of specialists. Pediatricians, dental specialists, surgeons, speech therapist, and psychologists must systematically and comprehensively plan treatment and rehabilitation. The palate may be repaired surgically or covered by an artificial device that closes or blocks the opening. Speech and language development need to be assisted by a speech therapist during the preschool years.
Genetic counseling is recommended for patients and their families.
Table 2. Treatment of manifestations in individuals with Simpson-Golabi-Behmel syndrome type 1
| Manifestation/Concern | Treatment | Considerations/Other |
|---|---|---|
| Macroglossia, micrognathia, &/or glossoptosis | Prompt standard treatment to maintain a secure airway; consider feeding/swallowing evaluation. | May require care of a craniofacial team |
| Cleft palate or bifid uvula | Assessment of feeding & management by a cleft/craniofacial team | |
| Obstructive sleep apnea (OSA) | If due to macroglossia, consider management similar to Beckwith-Wiedemann syndrome. | Limited data are available on prevalence & treatment of OSA in individuals w/SGBS1. |
| Sleep study & potential CPAP or hemiglossectomy, if indicated | ||
| Feeding difficulties | Milder feeding issues may be managed w/special nipples or nasogastric feeding in consultation w/specialist. | Limited data are available on treatment of feeding difficulties in individuals w/SGBS1. |
| Gastrostomy tube may be considered in those w/severe feeding issues. | ||
| Eyes | Standard treatment for strabismus and cataracts | |
| Hearing | Standard treatment for hearing loss | |
| Congenital heart defects & conduction abnormalities | Standard treatment as per cardiologist | |
| >Hypospadias/cryptorchidism in males | Standard treatment as per urologist | |
| Musculoskeletal findings (i.e., scoliosis) | Standard treatment as per orthopedist | |
| Seizure disorder | Standard treatment as per neurologist | |
| Hypoglycemia or suspected hyperinsulinism | Prompt treatment as per endocrinologist | Consider referral to tertiary care center for hyperinsulinism evaluation, if suspected. |
| Developmental delay | Early referral for developmental support/special education, which may incl physical therapy, occupational therapy, speech therapy, &/or cognitive therapy | Consider referral to a neurodevelopmental specialist &/or neuropsychiatric testing. |
| Cancer predisposition | For Wilms tumor, nephron sparing surgery should be considered, if possible. | See Surveillance for the recommended tumor-screening protocol. |
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years
- In the US, an IEP based on the individual’s level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
- Discussion about transition plans including financial, vocation/employment, and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual’s needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. Developmental Disabilities Administration is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction. Physical therapy is recommended to maximize mobility.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction. Assuming that the individual is safe to eat by mouth, feeding therapy – typically from an occupational or speech therapist – is recommended for affected individuals who have difficulty feeding due to poor oral motor control.
Communication issues. Consider evaluation for alternative means of communication (e.g., Augmentative and Alternative Communication [AAC]) for individuals who have expressive language difficulties.
Screening
Patients with Simpson Golabi Behmel syndrome type 1 need to be screened regularly for tumors. Screening should be done every three months until they are four years old. From ages four to seven years old, screening should be done every four months. After age seven, screening should be done every 6 months. Screening should include abdominal ultrasounds, blood tests, urine tests, and chest x-rays.
Little information on tumor risk in heterozygous females is available; there are currently only two reports of tumors in females with Simpson Golabi Behmel syndrome type 1. However, screening can be considered in affected females.
Table 3. Recommended surveillance for Males with Simpson-Golabi-Behmel syndrome type 1
| System/Manifestation | Evaluation | Frequency/Comment |
|---|---|---|
| Eyes | Ophthalmologic evaluation | Annually in childhood or as indicated |
| Hearing | Audiologic evaluation | Annually in childhood or as indicated |
| Respiratory | Sleep study | If history of sleep disturbance |
| Renal | Routine monitoring of renal function | If renal anomalies present |
| Musculoskeletal | Evaluate for scoliosis | At least annually or during periods of rapid growth |
| Endocrine | Monitor serum glucose levels for hypoglycemia secondary to increased risk for hyperinsulinemia. | Neonatal period |
| Neurodevelopment | Monitor for developmental progress. | At each clinic visit |
| Cancer predisposition | Tumor screening for Wilms tumors & hepatoblastomas | Abdominal ultrasound & serum AFP level every 3 mos from time of diagnosis until age 4 years |
| Renal ultrasound every 3 mos from age 4-7 years | ||
| Tumor screening for neuroblastoma & gonadoblastoma | Insufficient data to determine utility of screening in individuals with Simpson Golabi Behmel syndrome type 1 | |
| Follow up w/cancer predisposition specialist & physical examination | Every 6 months 6 |
Simpson Golabi Behmel syndrome prognosis
The spectrum of signs and symptoms associated with Simpson-Golabi-Behmel syndrome is very broad, varying from very mild forms in carrier females to infantile lethal forms in affected males. As many as 50% of affected males die in the newborn period, although the causes of this high mortality remain unknown 7; it has been suggested that it is probably related to cardiac abnormalities 4. People with milder cases often live into adulthood 1. Because of the varying degrees of manifestations and severity associated with the condition, prediction of prognosis and life expectancy most likely varies on an individual basis.
References- Simpson Golabi Behmel syndrome. https://ghr.nlm.nih.gov/condition/simpson-golabi-behmel-syndrome
- Tenorio J, Arias P, Martínez-Glez V, et al. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis. 2014;9:138. Published 2014 Sep 20. doi:10.1186/s13023-014-0138-0 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4254265
- Sajorda BJ, Gonzalez-Gandolfi CX, Hathaway ER, et al. Simpson-Golabi-Behmel Syndrome Type 1. 2006 Dec 19 [Updated 2018 Nov 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1219
- Garcia-Minaur S, Lapunzina Badia P, Martinez-Glen V, Nevado Blanco J, Santos Simarro F, Antonio Tenorio Castano J. Simpson-Golabi-Behmel syndrome. Orphanet. April 2015; https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=373
- Simpson-Golabi-Behmel Syndrome. https://rarediseases.org/rare-diseases/simpson-dysmorphia-syndrome
- Kalish JM, Doros L, Helman LJ, Henneka RC, Kuiper RP, Maas SM, Maher ER, Nichols KE, Plon SE, Porter CC, Rednam S, Schultz KAP, States LJ, Tomlinson GE, Zelley K, Druley TE. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. 2017;23:e115–22.
- Neri, G., Gurrieri, F., Zanni, G. and Lin, A. (1998), Clinical and molecular aspects of the Simpson‐Golabi‐Behmel syndrome. Am. J. Med. Genet., 79: 279-283. doi:10.1002/(SICI)1096-8628(19981002)79:4<279::AID-AJMG9>3.0.CO;2-H





{kind=link}