
Tetra amelia syndrome
Tetra-amelia syndrome is a very rare disorder characterized by the absence of all four limbs. “Tetra” is the Greek word for “four,” and “amelia” refers to the failure of an arm or leg to develop before birth. Tetra amelia syndrome can also cause severe malformations of other parts of the body, including the face and head, heart, nervous system, skeleton, and genitalia. The lungs are underdeveloped in many cases, which makes breathing difficult or impossible. Because children with tetra-amelia syndrome have such serious medical problems, most are stillborn or die shortly after birth. Tetra-amelia syndrome has been reported in only a few families worldwide 1.
Tetra amelia syndrome has been associated with a mutation in the WNT3 gene in one family and it appears to be inherited in an autosomal recessive manner 1. Treatment for those that survive depends upon the presence and severity of the associated symptoms and may require the coordinated efforts of a team of specialists 2.
Tetra amelia syndrome causes
Researchers have found a mutation in the WNT3 gene in people with tetra-amelia syndrome from one large family. The WNT3 gene is part of a family of WNT genes that play critical roles in development before birth. The protein produced from the WNT3 gene is involved in the formation of the limbs and other body systems during embryonic development. Mutations in the WNT3 gene prevent cells from producing functional WNT3 protein, which disrupts normal limb formation and leads to the other serious birth defects associated with tetra-amelia syndrome.
In other affected families, the cause of tetra-amelia syndrome has not been determined. Researchers believe that unidentified mutations in WNT3 or other genes involved in limb development are probably responsible for the disorder in these cases.
Tetra amelia syndrome inheritance pattern
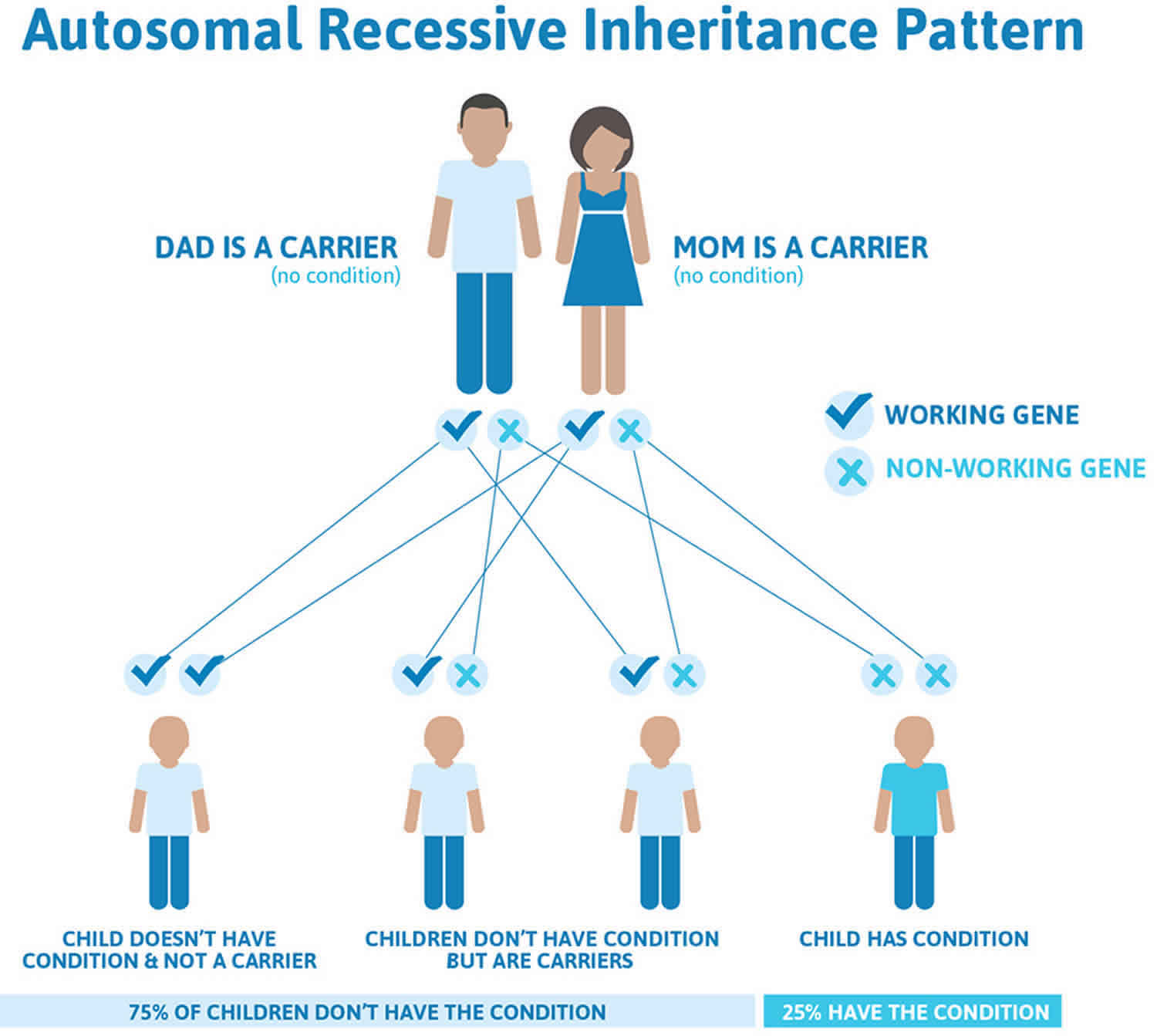
In most of the families reported so far, tetra-amelia syndrome appears to have an autosomal recessive pattern of inheritance. Autosomal recessive inheritance means both copies of the gene in each cell have mutations. The parents of an individual with tetra-amelia syndrome each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Tetra amelia syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Tetra amelia syndrome signs and symptoms
Tetra-amelia is characterized by the (complete) absence of all four limbs. In the few families described to date, tetra-amelia can also cause severe malformations of other parts of the body, including the face and head, heart, nervous system, skeleton, and genitalia. The lungs are underdeveloped in many cases, which makes breathing difficult or impossible. Because children with tetra-amelia syndrome have such serious medical problems, most are stillborn or die shortly after birth. Tetra-amelia syndrome has been reported in only a few families worldwide 1.
The following list is based on the findings in the affected individuals in the few families reported 3.
While the affected individuals in these families have all had similar findings, a mutation in WNT3 was only identified in the family reported by Niemann et al 4. With the exception of the family reported by Krahn et al 5 and Sousa et al 3, no molecular studies were undertaken in the other families. Therefore, in the absence of molecular genetic information to suggest subtypes of tetra-amelia syndrome, all reported cases are grouped together. The findings in the family with a WNT3 pathogenic variant are compared in Table 1 with the findings in families in which no molecular studies were undertaken or no WNT3 pathogenic variant was identified.
Craniofacial
- Eyes. Microphthalmia, cataract, microcornea, coloboma, palpebral fusion
- Ears. Absence of external ears (microtia), low-set ears
- Nose. Single naris, choanal atresia, prominent nose, absence of nose
- Mouth. Cleft lip/cleft palate, high and narrow palate, macrostomia, micrognathia
Urogenital
- Agenesis of kidney
- Rudimentary ovary and salpinx
- Persistence of cloaca
- Atresia of vagina
- Atresia of anus
- Atresia of urethra
- Hypospadias
- Absence of external genitalia
- Ambiguous genitalia
- Absence of scrotum
- Intra-abdominal location of testis
Cardiopulmonary
- Hypoplasia/aplasia of lungs, bilobular right lung
- Hypoplasia/aplasia of pulmonary vessels
- Diaphragmatic defect
- Ventricular septal defect
- Small right heart
- Mitral valve aplasia
Skeletal
- Hypoplasia/absence of pelvic bones
- Absence of vertebra
- Absence of rib
Central nervous system
- Agenesis of olfactory nerves
- Agenesis of optic nerves
- Agenesis of corpus callosum
- Hydrocephalus
Other
- Polyhydramnios
- Absence of nipples
- Gastroschisis
- Agenesis of suprarenal gland
- Agenesis of spleen.
Table 1. Clinical and autopsy findings in families with Tetra amelia syndrome
| Finding | Study | |||||||
|---|---|---|---|---|---|---|---|---|
| Zimmer et al 6, Gershoni-Baruch et al 7 | Rosenak et al 8 | Zlotogora et al 9 | Başaran et al 10 | Niemann et al 4 | Krahn et al 5 | Sousa et al 3 | Ragavan et al 11 | |
| Tetra-amelia | + | + | + | + | + | + | + | + |
| Cleft lip/ cleft palate | + | + | + | + | + | + | + | – |
| Micrognathia | − | + | − | + | − | + | + | – |
| Ear malformation | Absent | + | − | − | − | − | − | − |
| Eye malformation | + | − | − | + | + | − | − | − |
| Nose malformation | Absent | − | − | − | + | − | − | − |
| Mouth malformation | + | − | − | − | – | − | − | − |
| Heart malformation | − | − | ? | + | − | − | + | − |
| Pulmonary defects | + | Hypoplasia/ aplasia | ? | Aplasia | + | + | Aplasia | Hypoplasia |
| Pulmonary arteries | − | Hypoplasia | ? | ? | − | ? | Aplasia | − |
| Diaphragmatic defect | − | − | ? | − | + | − | − | − |
| Pelvic bones | Hypoplasia / aplasia | − | ? | − | Hypoplasia | − | − | − |
| Other skeletal defects | Absent vertebrae & ribs | − | ? | − | − | − | − | − |
| Renal malformation | − | − | ? | − | Agenesis | − | − | − |
| Genital malformation | + | – | ? | + | + | − | − | + |
| Anal atresia | + | − | − | − | + | − | − | − |
| Polyhydramnios | + | − | − | − | − | − | − | − |
| Hydrocephalus | + | + | ? | − | − | − | − | − |
| Other CNS defects | Agenesis of olfactory & optic nerves, corpus callosum | − | ? | − | − | − | − | − |
| Cases – total/autopsied | 7/2 | 3/2 | 5/0 | 2/1 | 4/3 | 2/2 | 1/1 | 1/1 |
Tetra amelia syndrome diagnosis
The diagnosis of tetra-amelia syndrome can be established clinically and is usually made on routine prenatal ultrasonography. WNT3 is the only gene in which pathogenic variants are known to cause tetra-amelia syndrome. The variant detection frequency is unknown as only a limited number of families have been studied.
Tetra amelia syndrome treatment
Affected infants are often stillborn or die shortly after birth. Management of (as yet unreported) persons who survive will depend on the presence and severity of associated malformations and require the support of several medical disciplines 2.
In nearly all reported cases, the pregnancy was terminated on diagnosis of tetra-amelia syndrome or infants died shortly after birth as a consequence of other malformations including pulmonary hypoplasia. Data on the management of tetra-amelia syndrome therefore do not exist.
It should be noted that (complete) absence of all extremities is principally not incompatible with life. Persons without extremities depend on extensive, life-long assistance with most daily activities. They would require specifically designed wheelchairs with assistive electronic technology and input control devices operated by head, chin, or tongue movements. Other individualized ambulatory devices may be indicated.
Tetra amelia syndrome prognosis
Data on the course of Tetra amelia syndrome or the prognosis are not available because the condition is rare. In nearly all reported cases, the pregnancy was terminated on diagnosis of tetra-amelia syndrome, or infants died shortly after birth as a consequence of other malformations such as pulmonary hypoplasia. Limb agenesis is generally compatible with life if adequate assistance is provided. The natural history of the disease is likely to be determined by extent and degree of associated manifestations.
References- Tetra amelia syndrome. https://ghr.nlm.nih.gov/condition/tetra-amelia-syndrome
- Niemann S. Tetra-Amelia Syndrome – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2007 Aug 28 [Updated 2012 Aug 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1276
- Sousa SB, Pina R, Ramos L, et al. Tetra-amelia and lung hypo/aplasia syndrome: new case report and review. Am J Med Genet A. 2008;146A(21):2799-2803. doi:10.1002/ajmg.a.32489
- Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U, Niswander L, Weber JL, Muller U. Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet. 2004;74:558–63.
- Krahn M, Julia S, Sigaudy S, Liprandi A, Bernard R, Gonnet K, Heuertz S, Bonaventure J, Chau C, Fredouille C, Levy N, Philip N. Tetra-amelia and lung aplasia syndrome: report of a new family and exclusion of candidate genes. Clin Genet. 2005;68:558–60.
- Zimmer EZ, Taub E, Sova Y, Divon MY, Pery M, Peretz BA. Tetra-amelia with multiple malformations in six male fetuses of one kindred. Eur J Pediatr. 1985;144:412–4.
- Gershoni-Baruch R, Drugan A, Bronshtein M, Zimmer EZ. Roberts syndrome or “X-linked amelia?”. Am J Med Genet. 1990;37:569–72.
- Rosenak D, Ariel I, Arnon J, Diamant YZ, Ben Chetrit A, Nadjari M, Zilberman R, Yaffe H, Cohen T, Ornoy A. Recurrent tetraamelia and pulmonary hypoplasia with multiple malformations in sibs. Am J Med Genet. 1991;38:25–8.
- Zlotogora J, Sagi M, Shabany YO, Jarallah RY. Syndrome of tetraamelia with pulmonary hypoplasia. Am J Med Genet. 1993;47:570–1.
- Başaran S, Yuksel A, Ermis H, Kuseyri F, Agan M, Yuksel-Apak M. Tetra-amelia, lung hypo-/aplasia, cleft lip-palate, and heart defect: a new syndrome? Am J Med Genet. 1994;51:77–80.
- Ragavan M, Reddy S, Kumar C. Tetra-amelia with lung hypoplasia and facial clefts, Roberts-SC syndrome: report of two cases. Pediatr Surg Int. 2010;26:1049–52.





{kind=link}