Sly syndrome
Sly syndrome also known as mucopolysaccharidosis type VII, is an extremely rare progressive inborn error of metabolism that affects many parts of the body. The clinical features of Sly syndrome vary from patient to patient, but all have short stature due to growth retardation and skeletal abnormalities, changes in bones visible on X-rays (dysostosis multiplex), and some degree of intellectual disability. Survival into adulthood is common for patients with milder cases and osteoarthritis is a common complication. Sly syndrome is inherited as an autosomal recessive genetic condition. The exact incidence of Sly syndrome is unknown, although it is estimated to occur in 1 in 250,000 newborns 1. Fewer than 100 cases have been reported in the United States. Males and females are affected in equal numbers. Sly syndrome is one of the rarest types of mucopolysaccharidosis.
The most severe cases of Sly syndrome are characterized by hydrops fetalis, a condition in which excess fluid builds up in the body before birth. Most babies with hydrops fetalis are stillborn or die soon after birth. Other people with Sly syndrome typically begin to show signs and symptoms of the condition during early childhood. The features of Sly syndrome include a large head (macrocephaly), a buildup of fluid in the brain (hydrocephalus), distinctive-looking facial features that are described as “coarse,” and a large tongue (macroglossia). Affected individuals also frequently develop an enlarged liver and spleen (hepatosplenomegaly), heart valve abnormalities, and a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia). The airway may become narrow in some people with Sly syndrome, leading to frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea). The clear covering of the eye (cornea) becomes cloudy, which can cause significant vision loss. People with Sly syndrome may also have recurrent ear infections and hearing loss. Affected individuals may have developmental delay and progressive intellectual disability, although intelligence is unaffected in some people with this condition.
Sly syndrome causes various skeletal abnormalities that become more pronounced with age, including short stature and joint deformities (contractures) that affect mobility. Individuals with this condition may also have dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Carpal tunnel syndrome develops in many children with Sly syndrome and is characterized by numbness, tingling, and weakness in the hands and fingers. People with Sly syndrome may develop a narrowing of the spinal canal (spinal stenosis) in the neck, which can compress and damage the spinal cord.
The life expectancy of individuals with Sly syndrome depends on the severity of symptoms. Some affected individuals do not survive infancy, while others may live into adolescence or adulthood. Heart disease and airway obstruction are major causes of death in people with Sly syndrome.
Figure 1. Sly syndrome
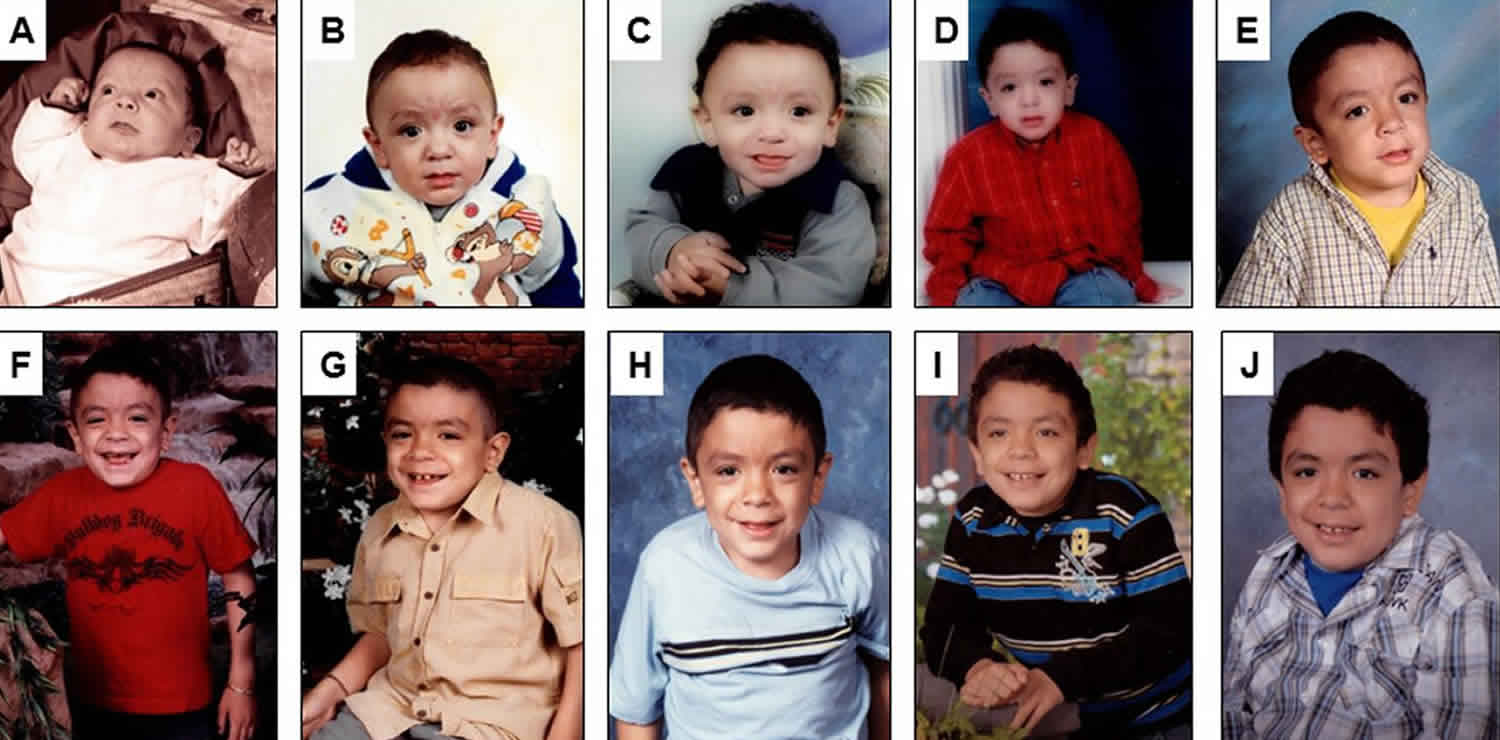
Footnote: Serial pictures of a male patient who survived a stormy early course, beginning with neonatal hydrops and subsequently progressed more slowly with cognitive impairment, hepatosplenomegaly, obstructive airway disease, heart valve abnormalities and dysostosis multiplex including progressive hip dysplasia. This patient died undergoing anaesthesia from a dental procedure at age 12 years, which is a complication from which these patients often suffer. (A) 2.5 months old, (B) 6 months old, (C) 1 year old, (D) 3 years old, (E) 5 years old, (F) 6 years old, (G) 7 years old, (H) 8 years old, (I) 9 years old and (J) 11 years old. X-rays are shown below in the section clinical course of the disease (Figures 2 and 3).
[Source 2 ]Figure 2. Sly syndrome spine X-rays
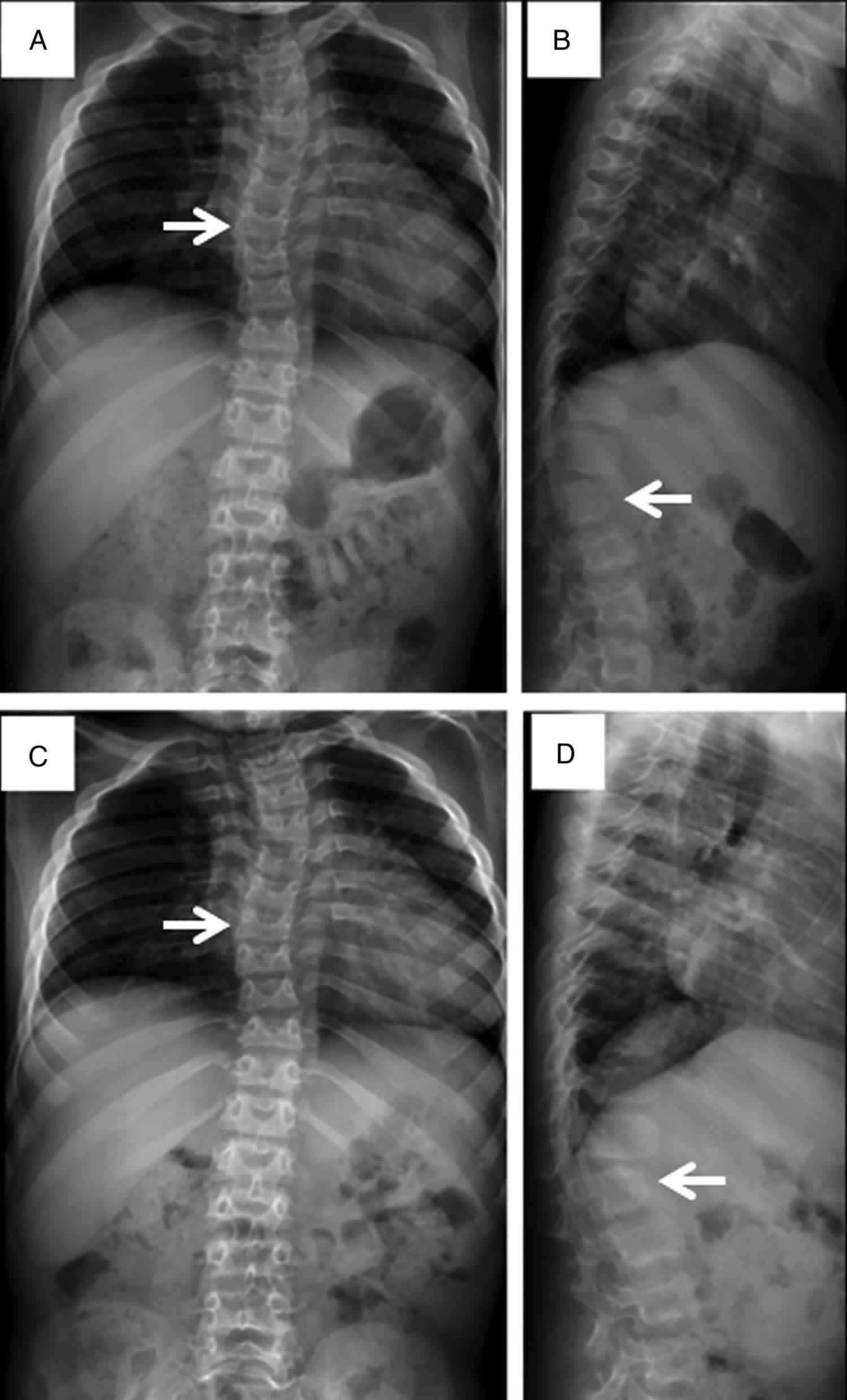
Footnote: Spine films in a patient with Sly syndrome (mucopolysaccharidosis VII) demonstrate findings common to all forms of mucopolysaccharidosis at age 7 years (A and B) and age 8 years (C and D). Anteroposterior views (A and C) reveal mild scoliosis (arrow) and broad ribs. The lateral films (B and D) demonstrate irregular anterior vertebral body growth, particularly at the L2 level (arrow).
[Source 2 ]Figure 3. Sly syndrome hip dysplasia
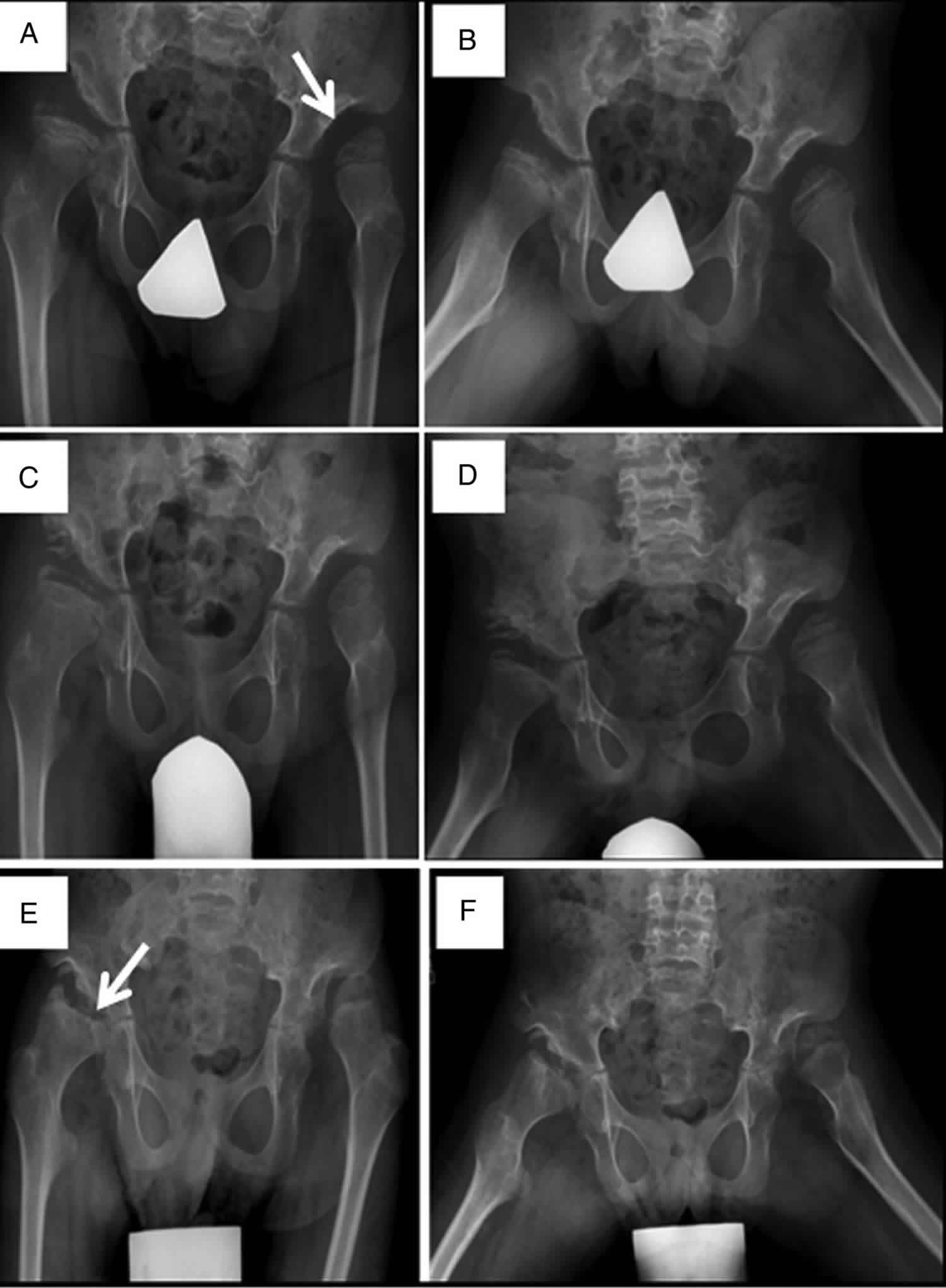
Footnote: Progressive hip dysplasia is seen in this patient with Sly syndrome (mucopolysaccharidosis VII). At age 7 years (A), the right hip shows subluxation with poor coverage, while the left hip is already dislocated and sits in a pseudoacetabulum (arrow). By age 8 years (C), an acetabular shelf procedure has been performed and by age 10 years (E), has not prevented further subluxation and femoral head erosion (arrow).
[Source 2 ]Sly syndrome causes
Mutations in the GUSB gene cause Sly syndrome. This gene provides instructions for producing the beta-glucuronidase (β-glucuronidase) enzyme, which is involved in the breakdown of large sugar molecules called glycosaminoglycans (GAGs). Glycosaminoglycans were originally called mucopolysaccharides, which is where this condition gets its name. Mutations in the GUSB gene reduce or completely eliminate the function of β-glucuronidase. The shortage (deficiency) of β-glucuronidase leads to the accumulation of glycosaminoglycans within cells, specifically inside the lysosomes. Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions such as Sly syndrome that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. The accumulation of glycosaminoglycans increases the size of the lysosomes, which is why many tissues and organs are enlarged in this disorder. Researchers believe that the glycosaminoglycans may also interfere with the functions of other proteins inside the lysosomes and disrupt many normal functions of cells.
Sly syndrome inheritance pattern
Sly syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
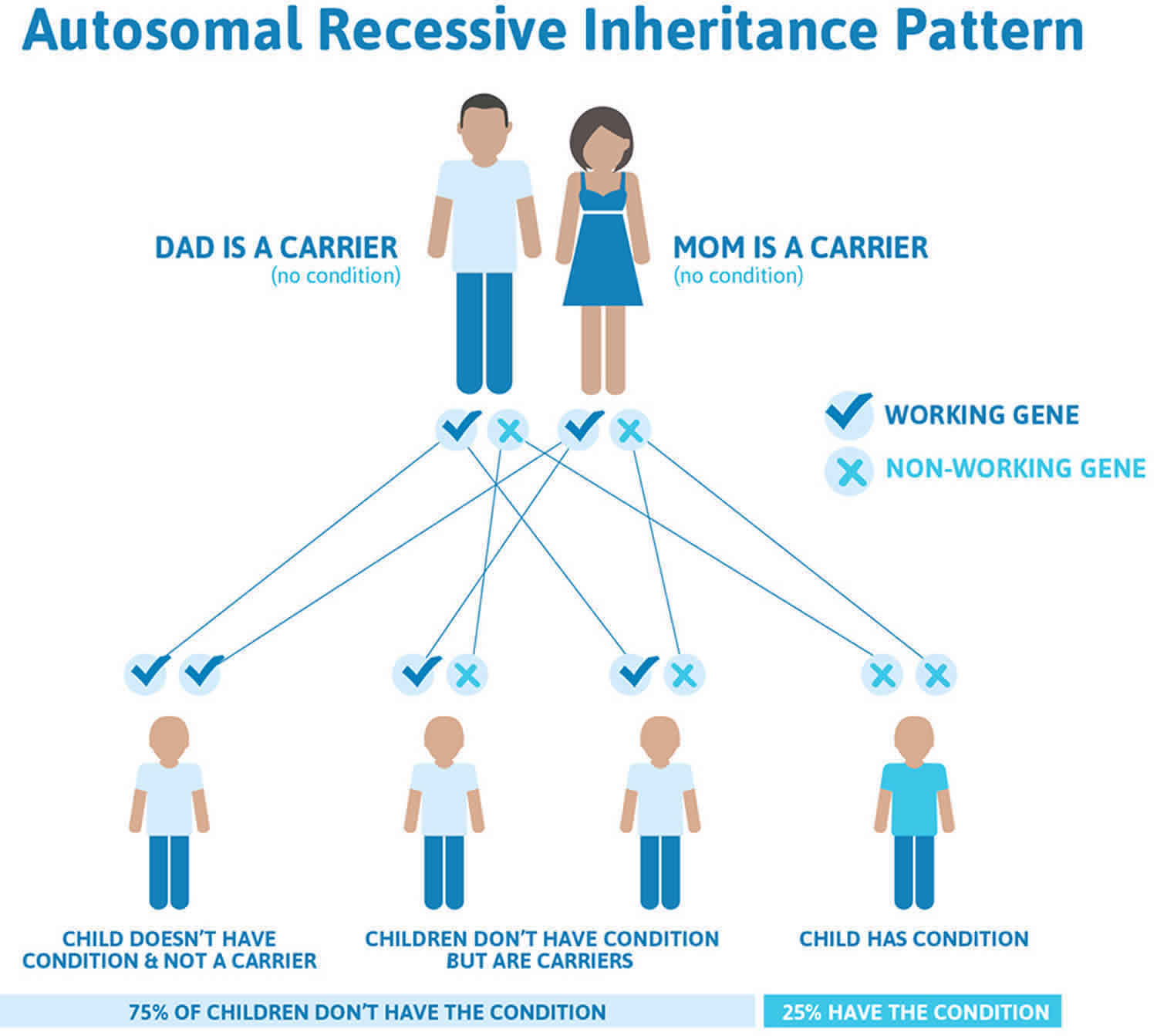
Figure 4 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 4. Sly syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Sly syndrome symptoms
Sly syndrome signs and symptoms are extremely variable. The most severe cases of Sly syndrome are characterized by hydrops fetalis, or when excess fluid accumulates in the body before birth. This can result in a stillborn or death shortly after birth. Neonatal jaundice or the yellowing of the skin may occur.
Children with more mild cases of Sly syndrome begin to show symptoms in early childhood. Children with Sly syndrome may have an unusually short trunk and growth disability, resulting in short stature (short trunk dwarfism). The head may be excessively large (macrocephalic) and the neck may be short. A variety of multiple bone deformities (dysostosis multiplex), which are frequently observed in people with mucopolysaccharidoses, are also common in children with Sly syndrome. These bone deformities may include a prominent breast bone (pectus carinatum), flared ribs, frequent hip dislocations, “frozen” joints (contractures), club foot, and/or an inward curve of the knees and outward bowing of the ankles (genu valgum).Rarely, spinal malformations may be present including mild curvature of the spine from side to side (scoliosis) and/or front to back (kyphosis). Children with this disorder may develop an unusual “coarse” facial appearance. Between the ages of 7 months and 8 years, cloudiness (opacity) may occur in the corneas of the eyes.
Affected individuals may have developmental delays in language and speech and progressive intellectual disability, although intelligence is normal in some people with this condition.
Other symptoms of Sly syndrome may include a swollen abdomen due to abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) and protrusion of the intestines through an abnormal opening in the muscular wall of the abdomen (inguinal hernia). In some children, the intestines may also protrude through the abdominal wall in the area of the navel (umbilical hernia). Some affected children may experience profound hearing loss, recurrent upper respiratory and middle ear infections, thickening of the soft tissues of the throat and/or vocal cords, an abnormally enlarged tongue (macroglossia), and/or heart problems (i.e., heart murmur or aortic regurgitation). Excessive hairiness (hirsutism) may be present.
Children may develop carpal tunnel syndrome characterized by numbness, tingling and weakness in the hands and fingers.
A mild form of Sly syndrome, beginning during the 2nd decade of life, has been described in the medical literature. In these patients, the symptoms of the disorder appear to be less severe than classical Sly syndrome and may include minor bony changes and very mild facial coarseness. Growth rate and mental abilities are usually normal. Abnormal enlargement of the liver and spleen has not been noticed in this form of Sly syndrome.
Sly syndrome diagnosis
Sly syndrome diagnosis is supported by x-ray evidence of dysostosis multiplex and detection of increased levels of urinary glycosaminoglycan (either chondroitin sulfate alone or chondroitin sulfate. dermatan sulfate and heparan sulfate) excretion, although this sign may be absent in adult forms. Diagnosis is confirmed by demonstration of beta-D-glucuronidase deficiency in cultured leucocytes or fibroblasts. Pseudodeficient alleles make mild forms more difficult to identify and prenatal diagnosis difficult. Molecular genetic testing for mutations in the GUSB gene is available to confirm the diagnosis. Prenatal diagnosis is possible through amniocentesis or chorionic villus sampling to measure beta-glucuronidase activity or molecular genetic testing for GUSB gene mutations.
Sly syndrome treatment
In 2017, Mepsevii (vestronidase alfa-jvbk) a recombinant human beta-glucuronidase, an enzyme replacement therapy, was approved to treat pediatric and adult patients with Sly syndrome. Mepsevii (vestronidase alfa-jvbk) has been shown to improve walking, lung function and hepatosplenomegaly in clinical trials 3. The patient was treated with intravenous infusions of 2 mg/kg of Mepsevii (recombinant human beta-glucuronidase) every other week. After 24 weeks of treatment, no serious adverse effects were noted. The levels of glycosaminoglycans (GAGs) in the urine decreased approximately 60% from the baseline, spleen and liver size declined, and pulmonary function improved as evidenced by increased tolerance for off-ventilator challenges. Quality of life was improved and he regained the ability to attend school and to eat some food orally. This patient’s experimental treatment with Mepsevii (vestronidase alfa-jvbk) is still ongoing. Long term outcome data are not yet available on the Mepsevii (recombinant human beta-glucuronidase) treated patients. Additional formal clinical trials of recombinant human beta-glucuronidase on patients with Sly syndrome are underway in the USA and Europe.
Gene therapy has been shown to be effective by several investigators. Davidson’s group 4 showed that gene therapy directed to mice brain could reverse established cognitive impairment. Potentially promising results were also reported in an Sly syndrome dog using retroviral gene therapy 5.
Other treatment of Sly syndrome is symptomatic and supportive. Bone deformities and hernias may require surgical correction. Ocular and cardiovascular abnormalities may also be treated surgically. Patients with mucopolysaccharidosis disorders may be sensitive to anesthesia because of malformations in the airway or cervical spine; therefore, precautions should be taken prior to surgery. Genetic counseling is recommended for people with Sly syndrome and their families.
About 46% of the patients with the infantile or adolescent Sly syndrome had surgeries. The most common included: hernia repair, hip replacement and cervical fusion. Seventeen per cent of the patients were able to perform daily activities by themselves, but more often, some assistance was required 2. Almost half of the patients with the infantile or adolescent form of the disease (46%) were able to walk by themselves without assistance 2. However, most patients lost this ability with time due to hip dysplasia. Common causes of death included complications of hydrops fetalis with respiratory and renal failure, heart disease, decreased pulmonary function and/or obstructive airway disease, complication of hematopoietic stem cell therapy and aspiration 2.
Hematopoietic stem cell therapy
A Japanese patient underwent successful hematopoietic stem cell therapy at 12 years of age and after 2 years the levels of glucuronidase were stable, with improvement of motor functions (walking, riding, and taking bath alone), recurrent infections and snoring; neurological damage has been kept stable 6. A Mexican female patient with Sly syndrome with genotype p.P408S/p.P415L underwent bone marrow transplantation at 11 years of age and was reported to be doing well 2 years after the transplantation 7. There were more than 7 patients that underwent hematopoietic stem cell therapy 8. The limited results suggest that bone marrow transplantation can slow or even prevent further neurological complications, but has little to no effect on the skeletal disease unless it is performed in an early stage.
Sly syndrome life expectancy
Sly syndrome life expectancy depends on the severity of symptoms. Some affected individuals do not survive infancy, while others may live into adolescence or adulthood. Survival to age 19-20 years has been reported in mild cases. Life expectancy is reduced as a result of heart disease and frequent upper respiratory tract infections, neurodegenerative complications and abnormalities of the gastrointestinal tract.
Distribution of causes of death by age. Causes included 2:
- Prenatal to 1 year, (50%) complications of hydrops fetalis, respiratory failure and renal failure;
- Ages 1–5 years (5%), obstructive airway disease;
- Ages 5–10 years (15%), heart disease, complications of bone marrow transplantation and pulmonary failure;
- Ages 10–15 (5%), decreased pulmonary function;
- Over 15 years of age, heart disease, pulmonary failure and/or obstructive airway disease, and aspiration/heart attack.
- Mucopolysaccharidosis type VII. https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-vii
- Montaño AM, Lock-Hock N, Steiner RD, et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). Journal of Medical Genetics 2016;53:403-418. https://jmg.bmj.com/content/53/6/403
- Fox JE, Volpe L, Bullaro J, Kakkis ED, Sly WS. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab 2015;114:203–8. doi:10.1016/j.ymgme.2014.10.017
- Liu G, Chen YH, He X, Martins I, Heth JA, Chiorini JA, Davidson BL. Adeno-associated virus type 5 reduces learning deficits and restores glutamate receptor subunit levels in MPS VII mice CNS. Mol Ther 2007;15:242–7. doi:10.1038/sj.mt.6300016
- Mango RL, Xu L, Sands MS, Vogler C, Seiler G, Schwarz T, Haskins ME, Ponder KP. Neonatal retroviral vector-mediated hepatic gene therapy reduces bone, joint, and cartilage disease in mucopolysaccharidosis VII mice and dogs. Mol Genet Metab 2004;82:4–19. doi:10.1016/j.ymgme.2004.01.015
- Yamada, Y, Kato, K, Sukegawa, K, Tomatsu, S, Fukuda, S, Emura, S, Kojima, S, Matsuyama, T, Sly, WS, Kondo, N, Orii, T. Treatment of MPS VII (Sly disease) by allogenic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant. 1998;21:629-634
- Islam, MR, Vervoort, R, Lissens, W, Hoo, JJ, Valentino, LA, Sly, WS. Beta-Glucuronidase P408S, P415L mutations: evidence that both mutations combine to produce an MPS VII allele in certain Mexican patients. Hum. Genet. 1996;98, 281-4.
- Montaño, AM, Lock-Hock, N, Steiner, RD, et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016;53, 403-418.





{kind=link}