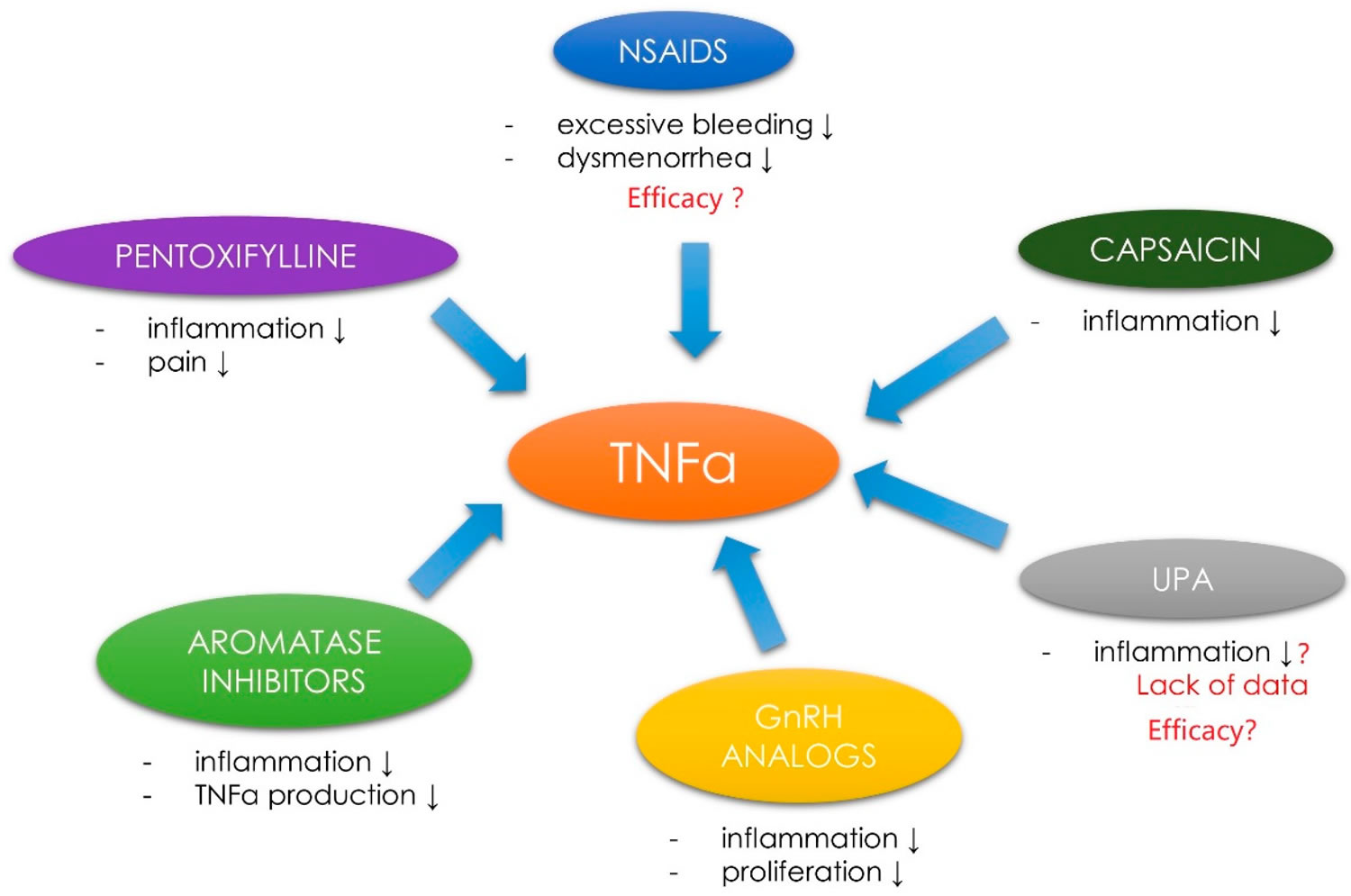
What is tumor necrosis factor
Tumor necrosis factor also known as tumor necrosis factor alpha (TNF-α), is a key pro-inflammatory cytokine (protein made by white blood cells in response to an antigen that causes the immune system to make a specific immune response), which can cause several inflammatory diseases or autoimmune diseases when inappropriately up-regulated 1. Tumor necrosis factor may boost a person’s immune response, and also may cause necrosis (cell death) of some types of tumor cells. Tumor necrosis factor was originally described as a circulating factor that can cause necrosis of tumors, but has since been identified as a key regulator of the inflammatory response. Tumor necrosis factor is being studied in the treatment of some types of cancer. Tumor necrosis factor inhibitor or anti tumor necrosis factor have been approved for human use in treating tumor necrosis factor-linked autoimmune diseases in the United States and other countries.
Tumor necrosis factor (TNF-α) was identified in 1975 as an endotoxin‐induced glycoprotein, which caused haemorrhagic necrosis of sarcomas that had been transplanted into mice 2. Human tumor necrosis factor was cloned in 1985 3 and recombinant tumor necrosis factor was shown to induce the hemorrhagic necrosis of transplanted methylcholanthrene‐induced sarcomas in syngeneic mice. Tumor necrosis factor alpha (TNF-α) has since been implicated in a diverse range of inflammatory, infectious and malignant conditions, and the importance of tumor necrosis factor in inflammation has been highlighted by the efficacy of anti‐tumor necrosis factor antibodies or administration of soluble tumor necrosis factor receptors (TNFRs) in controlling disease activity in rheumatoid arthritis and other inflammatory conditions.
Tumor necrosis factor is not usually detectable in healthy individuals, but elevated serum and tissue levels are found in inflammatory and infectious conditions 4 and serum levels correlate with the severity of infections 5. Although cells of the monocyte/macrophage lineage are the main source of tumor necrosis factor in inflammatory disease, a wide range of cells can produce tumor necrosis factor, including mast cells, T and B lymphocytes, natural killer (NK) cells, neutrophils, endothelial cells, smooth and cardiac muscle cells, fibroblasts and osteoclasts and involved in a wide range of pathological processes 6.
Tumor necrosis factor alpha (TNF-α) has been initially considered as a pro-inflammatory molecule 1. However, preclinical and clinical data have shown that it also mediates a paradoxical anti-inflammatory and immunomodulatory effect. Indeed, in laboratory mice models of type 1 diabetes or lupus nephritis, tumor necrosis factor may have a protective effect 7. Moreover, new onset or exacerbation of chronic inflammatory and autoimmune diseases has been observed in patients treated with anti-tumor necrosis factor therapies 8.
Tumor necrosis factor function
As a cytokine, tumor necrosis factor is involved with the inflammatory and immune response and can bind to TNF receptor 1 (TNFR1) or TNF receptor 2 (TNFR2) 9. Tumor necrosis factor occurs in numerous forms, both monomeric and trimeric, as well as soluble and transmembrane. Upon binding, tumor necrosis factor triggers the activation of numerous pathways including the NFkB and MAPK pathways. This leads to the production of numerous inflammatory cytokines and can also lead to tumor necrosis factor-induced apoptotic pathway initiation.
The inflammatory cytokine tumor necrosis factor alpha (TNF-alpha) is expressed in immune and nonimmune cell types including macrophages, T cells, mast cells, granulocytes, natural killer (NK) cells, fibroblasts, neurons, keratinocytes and smooth muscle cells as a response to tissue injury or upon immune responses to pathogenic stimuli.
Tumor necrosis factor is produced predominantly by activated macrophages and T-lymphocytes as a 26 kDa protein, pro‐tumor necrosis factor, which is expressed on the plasma membrane, where it can be cleaved in the extracellular domain by the matrix metalloproteinases, which result in the release a soluble 17 kDA soluble form. Both membrane‐associated and soluble tumor necrosis factors are active in their trimeric forms, and the two forms of tumor necrosis factor may have distinct biological activities. Tumor necrosis factor alpha converting enzyme (TACE, also known as ADAM‐17) mediates release of tumor necrosis factor from the cell surface 10, but is involved in processing several cell‐membrane associated proteins, including tumor necrosis factor receptors, which are released by its action to produce soluble froms that can neutralize the actions of tumor necrosis factor 11. Tumor necrosis factor alpha converting enzyme (TACE) may therefore be either pro‐ or anti‐inflammatory, depending on whether it acts on an effector (eg macrophage) or target (eg endothelial) cell, releasing ligand or receptors, respectively.
Tumor necrosis factor alpha (TNF-α) is also found in smooth muscle cells as a response to tissue injury or upon immune responses to various stimuli 12. Tumor necrosis factor-α uses two types of receptors: tumor necrosis factor-α receptor type 1 (TNFR1) (also known as TNFRSF1A, CD120a, p55) and tumor necrosis factor type 2 (TNFR2) (also known as TNFRSF1B, CD120b, p75) (Figure 1). Tumor necrosis factor-α receptor type 1 (TNFR1) is expressed in most tissues, whereas tumor necrosis factor type 2 (TNFR2) is found primarily in the immune system cells 13. After binding to the receptor, the TNF-α molecule may activate one of the three potential effects: 1) activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which is a transcription factor involved in cell survival, proliferation and the inflammatory response [this include NF-κB-inducing kinase (NIK) 14 and I kappa B kinase (IKK) 15]; 2) activation of the mitogen-activated protein kinases (MAPK) pathways [through c-jun N-terminal kinase (JNK) 16 and on the other hand through receptor interacting protein (RIP) kinases family 17 and MAP kinase kinase (MEKK) 18], involved in cell differentiation and proliferation, and 3) induction of death signaling (Figure 1). Most of the mentioned pathways are tumor necrosis factor receptor-associated factor (TRAF)2 dependent as presented in Figure 1 19.
Tumor necrosis factor alpha (TNF-α) is a pleiotropic cytokine which has been identified as the key regulator of the inflammatory response 20. Tumor necrosis factor alpha (TNF-α) also plays a major role in the cell cycle, being the controller of growth, differentiation, and apoptosis 21. Tumor necrosis factor-α has multiple biological functions throughout the human body, including fever and acute phase stimulation, promotion of the adhesion molecule expression, phagocytosis stimulation, appetite suppression, and modulation of insulin resistance 13. Tumor necrosis factor-α can be an anti-neoplastic and anti-angiogenic agent which stimulates the immune system to fight cancer cells 22. It has been found that tumor necrosis factor-α is an important gene expression regulator. The interaction of chemokines and their receptors may cause the amplification of cellular signaling pathways and induce the expression of proteins responsible for proliferative cells or changes in their normal metabolism 23. Despite the anti-cancer properties of tumor necrosis factor-α, elevated levels of tumor necrosis factor-α are not always capable of destroying all abnormal cells and, paradoxically, they can cause severe symptoms related to tumor occurrence 24. Dysregulation of tumor necrosis factor-α production and distribution has been demonstrated in various human diseases, including cancers, dermatoses, and inflammatory bowel diseases (Table 1) 23.
Figure 1. Tumor necrosis factor alpha receptors
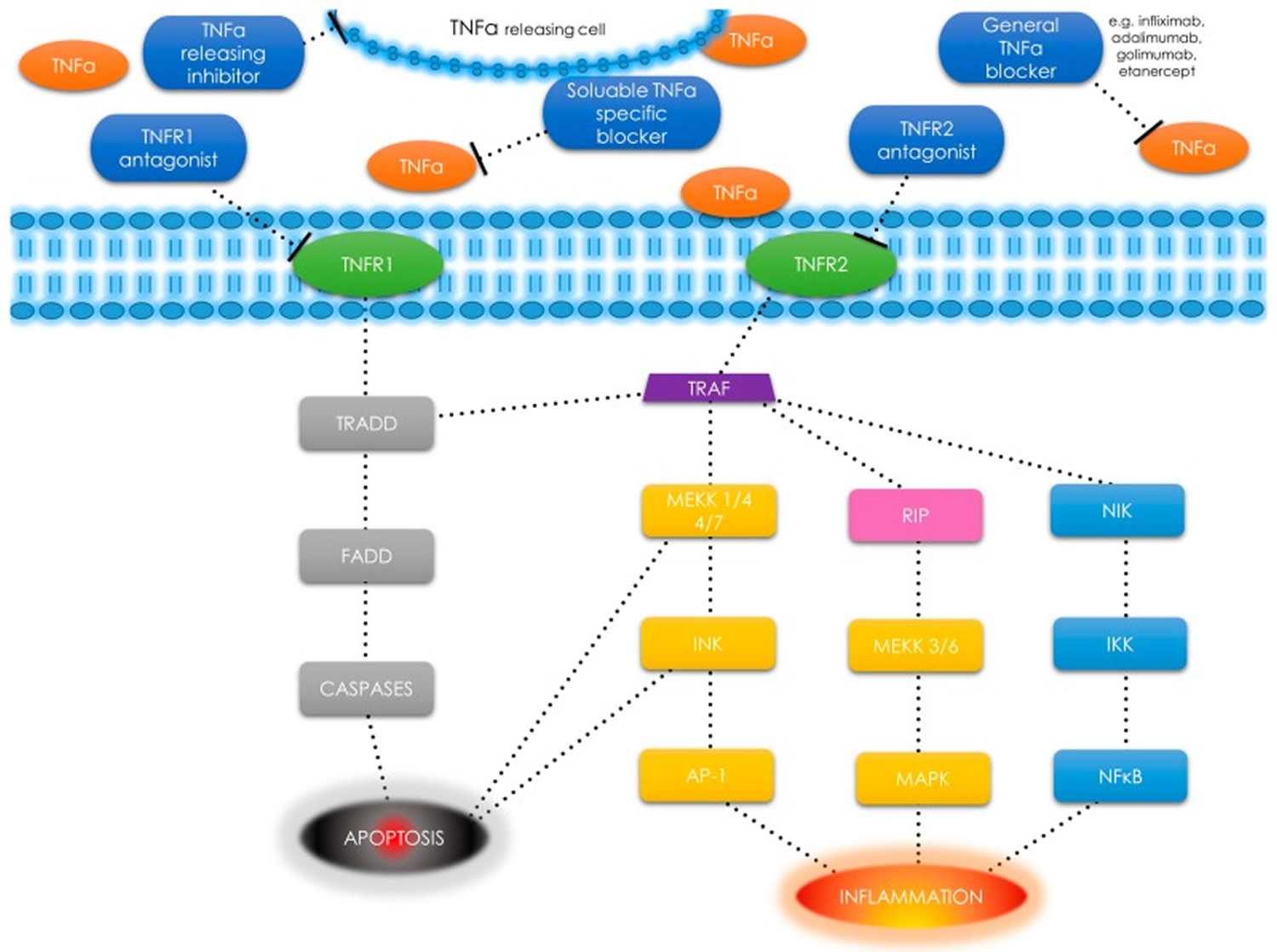
Footnote: TNF-α receptors, pathways and different types of signals—schematic diagram. TNF-α has an ability to induce apoptosis, cell survival or inflammation depending on selected pathway.
Abbreviations: Tumor necrosis factor α (TNF-α); tumor necrosis factor α receptor (TNFR); tumor necrosis factor receptor type 1-associated death domain (TRADD); Fas-associated protein with death domain (FADD); tumor necrosis factor receptor-associated factor (TRAF); mitogen-activated protein kinase kinase kinase (MEKK); c-jun N-terminal kinase (JNK); activator protein 1 (AP-1); receptor interacting protein (RIP); mitogen-activated protein kinases (MAPK); nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB); NF-κ-B-inducing kinase (NIK); I κB kinase (IKK).; T-bar as a drug – binding point interaction.
Table 1. Examples of human diseases with dysregulated tumor necrosis factor alpha (TNF-α) production
| Field | Examples |
|---|---|
| Rheumatology | Rheumatoid arthritis Psoriatic Arthritis Ankylosing Spondylitis |
| Dermatology | Plaque psoriasis |
| Ophtalmology | Uveitis |
| Psychiatry | Depression |
| Gastroenterology | Crohn’s Disease Ulcerative Colitis |
| Urology | Renal cell carcinoma |
| Gynecology | Ovarian cancer Uterine fibroids |
| Neurology | Alzheimer’s Disease |
Physiological roles of tumor necrosis factor
One of the major biological roles of tumor necrosis factor is in the host defence to bacterial, viral and parasitic infections. Physiologically, tumor necrosis factor is important for the normal response to infection, but inappropriate or excessive production can be harmful. tumor necrosis factor was originally identified as an endotoxin lipopolysaccharide (LPS)‐induced humoral mediator of murine cachexia, the syndrome of anorexia, weight loss and protein wasting that complicates cancer and chronic infection and inflammation 26. Tumor necrosis factor was confirmed as the principal mediator of the lethal effect of Escherichia coli‐derived endotoxin by demonstrating that passive immunization of mice with rabbit anti‐serum to tumor necrosis factor protected mice from its lethality 27. Baboons passively immunized with a neutralizing monoclonal anti‐tumor necrosis factor antibody and subsequently infused with a lethal dose of live E. coli were protected against shock, vital organ dysfunction, persistent stress hormone release and death 28. However, although a recombinant, soluble fusion protein that combines an extracellular portion of the human tumor necrosis factor receptor and the Fc portion of IgG (TNFR : Fc) neutralizes tumor necrosis factor and prevents death in animal models of bacteraemia and endotoxaemia, in patients with septic shock, treatment with tumor necrosis factor receptor and the Fc portion of IgG did not reduce mortality, and higher doses appeared to be associated with increased mortality 29.
The importance of tumor necrosis factor as a modulator of the host response to infection has been confirmed by studies using tumor necrosis factor receptor‐deficient mice. Mice deficient in TNFR1 are resistant to lethal dosages of either lipopolysaccharides or Staphylococcus aureus enterotoxin B, but have severely impaired clearance of the intracellular bacterium Listeria monocytogenes and readily succumb to this infection 30. The activity of tumor necrosis factor in endotoxin‐induced lethal shock and innate resistance to Listeria appears to be independent of TNFR2 31. tumor necrosis factor has since been shown to be essential for the formation and maintenance of granulomas, which limit dissemination of Listeria and other infections, including mycobacteria 32, and a number of granulomatous infections have been reported in association with the use of tumor necrosis factor antagonists to treat human inflammatory disease 33.
Evidence also supports a key role for tumor necrosis factor in parasitic and viral infections. tumor necrosis factor levels are increased in the serum of children with uncomplicated Plasmodium falciparum malaria, and markedly increased in children with a fatal outcome from cerebral malaria, leading to speculation that increased tumor necrosis factor production is a normal host response to P. falciparum infection, but that excessive levels of production may predispose to cerebral malaria and a fatal outcome 5.
Tumor necrosis factor appears to be central for the ICAM‐1‐dependent recruitment of mononuclear cells and microvascular damage that occurs in cerebral malaria 34. Anti‐tumor necrosis factor therapy inhibits fever in cerebral malaria 35 but does not improve survival 36. Support for a role of tumor necrosis factor in host defences against viruses has been provided by the existence of viruses encoding tumor necrosis factor‐binding proteins in their genome 37. Furthermore, side‐effects typically associated with viraemia, including fever, rigors, headache and fatigue, were observed in early trials of tumor necrosis factor in cancer patients 38. Although initially described as a tumouricidal agent, toxicity has limited the role of tumor necrosis factor as a chemotherapeutic agent for cancer. Furthermore, tumor necrosis factor may under some circumstances contribute to carcinogenesis by promoting proliferation, invasion and metastasis of tumour cells 39. However, isolated limb perfusion with tumor necrosis factor is of value in palliating patients with metastatic sarcoma and melanoma 40, providing tumour control and limb salvage for the short survival of patients, and isolated hepatic perfusion with tumor necrosis factor has been used in patients with hepatic metastases 41.
Although tumor necrosis factor blockade failed to be of benefit in severe sepsis, the trials that were undertaken paved the way for its use in chronic inflammatory diseases such as rheumatoid arthritis, which in turn has highlighted the physiological roles of tumor necrosis factor in sepsis and malignancy.
Diseases associated with tumor necrosis factor
Rheumatoid arthritis
Rheumatoid arthritis is a chronic autoimmune inflammatory disorder affecting approximately 1% of the population, characterized by inflammation of synovial tissue, leading to progressive damage, erosion of adjacent cartilage and bone and chronic disability. The inflammation is associated with accumulation of inflammatory cells, predominantly T cells and macrophages, but also B cells, plasma cells and dendritic cells. There is synovial hyperplasia and angiogenesis is a prominent feature 42. Many pro‐inflammatory cytokines, including IL‐1, IL‐6, tumor necrosis factor and GM‐CSF, are produced within the inflamed joint 43.
Support for a dominant role of tumor necrosis factor was provided by a number of in vitro and in vivo studies. Production of a range of pro‐inflammatory cytokines by cultured cells from the joints of patients with rheumatoid arthritis can be down‐regulated by a neutralizing antibody to tumor necrosis factor 85, 86. Neutralizing tumor necrosis factor with either anti‐tumor necrosis factor antibody or a recombinant soluble tumor necrosis factor receptor ameliorates joint disease in a murine model of collagen‐induced arthritis 44. Deleting tumor necrosis factor AU‐rich elements (AREs) from the mouse genome resulted in profound temporal and spatial deregulation of tumor necrosis factor production, characterized by the persistent accumulation and decreased rates of decay of the mutant tumor necrosis factor mRNA. This deregulation resulted in overexpression of tumor necrosis factor and the development of chronic inflammatory arthritis and inflammatory bowel disease 45.
An open Phase I/II clinical trial of a murine‐human chimeric neutralizing monoclonal antibody to tumor necrosis factor in 20 patients with active rheumatoid arthritis demonstrated that the treatment was safe and well tolerated and resulted in significant clinical and laboratory improvements 46. The efficacy of anti‐tumor necrosis factor treatments in rheumatoid arthritis was subsequently confirmed in randomized controlled trials, which established that methotrexate and anti‐tumor necrosis factor treatment have a synergistic effect, and demonstrated that long‐term treatment is feasible 47, although loss of response may occur in about half of patients during the first year 48.
Inflammatory bowel disease
tumor necrosis factor immunoreactivity is increased the lamina propria in intestinal specimens from patients with Crohn’s disease and ulcerative colitis 49, and mice overexpressing tumor necrosis factor develop a Crohn’s disease‐like inflammatory bowel disease 45.
Infliximab has been shown to be effective in inducing remission, and an effective maintenance therapy for patients with Crohn’s disease with and without fistulas 50. A randomized controlled trial demonstrated that the humanized anti‐tumor necrosis factor antibody CDP571 was an effective for treatment of patients with moderate to severe Crohn’s disease 51, but subsequent studies showed that CDP571 is not effective for long‐term treatment of unselected patients with moderate to severe Crohn’s disease 52.
A systemic review of randomized controlled trials of tumor necrosis factor‐blocking agents in patients with active Crohn’s disease found that infliximab and CDP571, but not etanercept, may be effective in inducing remission 53.
The role of tumor necrosis factorα‐blocking agents in ulcerative colitis is less clear, and recent studies have yielded conflicting results. A systematic review concluded that in patients with moderate to severe ulcerative colitis whose disease is refractory to conventional treatment using corticosteroids and/or immunosuppressive agents, infliximab is effective in inducing clinical remission, inducing clinical response, promoting mucosal healing and reducing the need for colectomy, at least in the short term 54.
Ankylosing spondylitis
Ankylosing spondylitis is an inflammatory arthritis that predominantly affects the spine and sacroiliac joints. It occurs more commonly in patients with Crohn’s disease. tumor necrosis factor has been detected in the sacro‐iliac joints of patients with ankylosing spondylitis 55, particularly in early active disease 56, and elevated serum levels of tumor necrosis factor correlate with disease activity 57.
A randomized, double‐blind trial of etanercept showed that treatment with etanercept for 4 months resulted in rapid, significant and sustained improvement in patients with ankylosing spondylitis 58. Subsequent studies have confirmed the efficacy and safety of etanercept in patients with active ankylosing spondlitis over 2 years of continuous treatment 59, and have shown that etanercept is effective in patients with early onset ankylosing spondylitis before the age of 60.
Infliximab is also an effective agent in patients with active ankylosing spondylitis, for inducing and maintaining remission and readministration to treat relapse after discontinuation of long‐term therapy 61.
Psoriasis
Psoriasis is an inflammatory skin disorder, in which an inflammatory cell infiltrate is associated with hyperkeratotic lesions, giving rise to typical psoriatic plaques. tumor necrosis factor,TNFR1 and TNFR2 are upregulated in dermal blood vessels in involved skin from patients with psoriasis 62.
Randomized trials showed that infliximab resulted in a rapid and significant improvement in psoriatic plaques 63. Up to one‐third of patients with psoriasis develop an inflammatory arthritis, which can affect both spinal and peripheral joints. Early studies suggested a role for etanercept in the treatment of psoriatic arthritis, and subsequent studies have shown that infliximab, etanercept and adalimumab are all effective treatments for the dermatological and articular manifestations of psoriasis 64.
Disease of the central nervous system
In the central nervous system, tumor necrosis factor is produced primarily by microglia and astrocytes in response to a wide range of pathological processes, including infection, inflammatory disease, ischaemia and traumatic injury 65. However, tumor necrosis factor has been shown to have both harmful and beneficial effects in the injured brain 66. Inhibition of tumor necrosis factor ameliorates ischaemic brain injury in mice 67, whereas mice lacking tumor necrosis factor are highly susceptible to experimental autoimmune encephalomyelitis, and treatment with tumor necrosis factor dramatically reduces disease severity 128. tumor necrosis factor‐mediated protection against experimental autoimmune encephalomyelitis does not require TNFR1, although TNFR1 appears to be necessary for detrimental effects of tumor necrosis factor, which occur during the acute phase of the disease 68. In contrast, TNFR2 has been shown to promote proliferation of oligodendrocyte progenitors and remyelination in a neurotoxicant murine model of demyelination 69. Neutralization of tumor necrosis factor failed to benefit patients with relapsing–remitting multiple sclerosis, and significantly increased exacerbations 70.
Cardiovascular disease
Tumor necrosis factor has also been implicated in the pathogenesis of a number of cardiovascular diseases, including atherosclerosis, myocardial infarction, heart failure, myocarditis and cardiac allograft rejection, and vascular endothelial cell responses to tumor necrosis factor may underlie the vascular pathology in many of these conditions. However, clinical trials have demonstrated no clinical benefit of tumor necrosis factor blockade in congestive cardiac failure. This may be because TNFR1 and TNFR2 differentially regulate cardiac responses to tumor necrosis factor. In transgenic mice with tumor necrosis factor‐induced cardiomyopathy, ablation of the TNFR2 gene exacerbates heart failure and reduces survival, whereas ablation of TNFR1 blunts heart failure and improves survival 71. In cardiac allografts either tumor necrosis factor receptor is capable of mediating a response that will culminate in graft arterial disease 72.
Patients with chronic inflammatory conditions such as rheumatoid arthritis have an increased incidence of cardiovascular disease. Inflammatory mediators, including tumor necrosis factor, have been implicated in this increased cardiovascular risk, and there is some evidence that anti‐tumor necrosis factor therapy ameliorates this risk in patients with rheumatoid arthritis 73.
Respiratory disease
Tumor necrosis factor has been implicated in the pathophysiology of many inflammatory lung diseases, including chronic bronchitis, chronic obstructive pulmonary disease, acute respiratory distress syndrome and asthma 74. In asthma, tumor necrosis factor has been implicated in airway inflammation and remodelling, and may play a role in bronchial hyper‐responsiveness. Leukocytes from bronchiolar lavage of asthma patients have increased release of tumor necrosis factor 75, and inhaled tumor necrosis factor increases airway responsiveness in normal subjects and is associated with a pulmonary neutrophil infiltration, assessed by induced sputum. To date there are no randomized controlled trials of tumor necrosis factor blockade in asthma, but patients with asthma who received infliximab for rheumatoid arthritis have demonstrated a significant improvement, and an open label uncontrolled study of etanercept in 17 subjects with severe asthma demonstrated an improvement in asthma symptoms, lung function and bronchial hyper‐responsiveness following 12 weeks of entanercept 76. A pilot study of patients with refractory asthma provided evidence for a role of tumor necrosis factor, and demonstrated a beneficial effects of etanercept on markers of asthma control 77.
Renal disease
Tumor necrosis factor has been implicated in the pathogenesis of many renal diseases, including ischaemic renal injury, renal transplant rejection and glomerulonephritis, which is often part of a systemic vasculitis. In diseases associated with renal inflammation, different forms of tumor necrosis factor blockade vary in their efficacy and adverse effects, and these differences may be attributed to different effects on signalling though tumor necrosis factor receptor subtypes. In acute renal injury, TNFR2‐mediated cell proliferation may be important for tubular cell regeneration 78, whereas in proliferative forms of glomerulonephritis, tumor necrosis factorR1‐mediated cytotoxicity and inhibition of TNFR2‐mediated cellular proliferation may be more desirable.
TNFR2 is essential for the development of renal injury in a model of immune complex glomerulonephritis induced by anti‐GBM antibody, raising the possibility that selective blockade of TNFR2 may be a promising strategy for treatment of immune‐mediated glomerulonephritis 79.
As discussed above, infliximab may be more effective at blocking signalling through TNFR2 than etanercept. In support of this, infliximab is an effective treatment for patients with refractory Wegener’s granulomatosis 80, in which lymphocyte activation and glomerular cell proliferation are important in pathogenesis. In contrast, etanercept is not effective for the maintenance of remission in patients with Wegener’s granulomatosis, and its use is associated with an increased incidence of solid tumours 81.
Other inflammatory diseases
As the potential role of tumor necrosis factor blockade has become clear, its successful use has been reported in an increasing number of inflammatory conditions. These include juvenile rheumatoid arthritis 82; therapy‐resistant sarcoidosis, a multisystemic disorder characterized histologically by the presence of granulomatous inflammation 83; inflammatory myopathies 84; Behcet disease 85; and inflammatory eye disease 86.
Tumor necrosis factor inhibitors
Tumor necrosis factor alpha inhibitors, e.g., infliximab, etanercept, adalimumab, certolizumab, and golimumab has been FDA approved for use in rheumatoid arthritis, ulcerative colitis, Crohn disease, ankylosing spondylitis, juvenile idiopathic arthritis, plaque psoriasis and, psoriatic arthritis. Adalimumab also has approval for hidradenitis suppurativa and uveitis. Non-FDA-approved indications include pyoderma gangrenosum, pustular psoriasis, and graft versus host disease (etanercept). Etanercept is a recombinant human soluble fusion protein in which TNFR2 is coupled to the Fc portion of IgG. Infliximab is a human–murine chimeric IgG1 monoclonal anti‐TNF antibody. Adalibumab is a human anti‐human TNF antibody produced by phage display. Other agents in clinical development have used PEGylation to couple a large molecular weight polyethylene glycol molecule to the tumor necrosis factor antagonist, with the aim of prolonging the half‐life. PEG–sTNFR1 is a pegylated form of soluble TNFR1, and CDP‐870 is a pegylated Fab of the humanized anti‐TNF antibody CDP‐571.
Different anti‐tumor necrosis factor therapies may have different binding and pharmacokinetic profiles. The chimeric monoclonal antibody infliximab may be a more potent inhibitor of TNFR2 signalling than the TNFR2–Fc fusion protein entarecept. Infliximab binds transmembrane TNF with higher avidity, forming more stable complexes and more effectively inhibiting the actions of transmembrane TNF than entarecept 87. Transmembrane TNF is superior to soluble TNF in activating TNFR2 in various systems, including T cell activation, thymocyte proliferation and granulocyte/macrophage colony‐stimulating factor production 88. Thus, under certain conditions infliximab may be more effective at blocking signalling through TNF2 than etanercept.
The tumor necrosis factor-alpha inhibitors, including etanercept, infliximab, adalimumab, certolizumab pegol, and golimumab, all bind to the cytokine tumor necrosis factor and inhibit its interaction with the TNF receptors 89. Etanercept is a fusion protein of two TNFR2 receptor extracellular domains and the Fc fragment of human IgG1 90. It inhibits the binding of both TNF-alpha and TNF-beta to cell surface TNFRs. Infliximab is a chimeric monoclonal antibody that includes a murine variable region and constant human region 91. It binds to the soluble and transmembrane forms of TNF-alpha and inhibits the binding of TNF-alpha to TNFR. Adalimumab and golimumab are fully human monoclonal antibodies against TNF-alpha and, like infliximab, these antibodies bind to TNF-alpha and inhibit its binding to TNFR. Certolizumab is a humanized Fab fragment conjugated to polyethylene glycol (PEG).
FDA-approved Indications (alphabetical list)
- Ankylosing spondylitis (etanercept, infliximab, adalimumab, certolizumab pegol, and golimumab)
- Crohn disease (infliximab, adalimumab and certolizumab pegol)
- Hidradenitis suppurativa (adalimumab)
- Juvenile idiopathic arthritis (adalimumab)
- Plaque psoriasis (etanercept, infliximab and adalimumab)
- Polyarticular juvenile idiopathic arthritis (etanercept)
- Psoriatic arthritis (etanercept, infliximab, adalimumab, certolizumab pegol, and golimumab)
- Rheumatoid arthritis (etanercept, infliximab, adalimumab, certolizumab pegol, and golimumab)
- Ulcerative colitis (infliximab, adalimumab and golimumab)
- Uveitis (adalimumab)
Off-label Indications
- Graft-vs-host disease
- Juvenile idiopathic arthritis
- Pustular psoriasis
- Pyoderma gangrenosum
Etanercept is administered subcutaneously. The injection sites should be rotated and given at least one inch apart from previous injection sites.
Infliximab is administered as an infusion. The infusion should occur over a period of 2 hours and should not be co-administered with other agents. Infusion-related reactions are possible, and if necessary, antihistamines, acetaminophen, and corticosteroids can be used to treat these reactions.
Adalimumab is administered by subcutaneous injection. The injection sites should be rotated and away from previous injection sites.
Certolizumab pegol is subcutaneously administered into the thigh or abdomen.
Golimumab can be administered intravenously (IV) or subcutaneously. Infusions should occur over a period of 30 minutes and should not be co-administered with any other agents. Subcutaneous injections are administered using an autoinjector.
Tumor necrosis factor inhibitor side effects
The following side effects were observed in greater than 10% of patients receiving a tumor necrosis factor-alpha, including etanercept, infliximab, adalimumab, certolizumab pegol and golimumab 92.
- Central Nervous System-related side effects: Headache
- Dermatology-related side effects: Skin rash
- Gastrointestinal-related side effects: Abdominal pain, nausea, diarrhea
- Hematology-related side effects: Anemia
- Hepatic-related side effects: Increased serum alanine aminotransferase (ALT)
- Immunology-related side effects: Increased ANA titer (~50%), antibody development
- Infection-related side effects: Infection, including serious infection and abscess
- Respiratory-related side effects: Upper respiratory tract infections, sinusitis, cough, pharyngitis
- Miscellaneous side effects: Infusion-related reaction, injection site reactions
- Other serious concerns related to side effects: Anaphylaxis and hypersensitivity reactions, autoimmune disorders, demyelinating CNS disease, heart failure, aplastic anemia, pancytopenia, reactivation of hepatitis B, leukopenia, neutropenia
US boxed warnings (for all TNF-alpha inhibitors):
- Malignancy: Malignancies, including lymphoma, have been reported in patients receiving tumor necrosis factor alpha inhibitors. The malignancies arose approximately 30 months (ranging from 1 month to 84 months) after the first dose of the tumor necrosis factor alpha inhibitor and many of the cases were in pediatric and young adult populations. Additionally, cases of hepatosplenic T-cell lymphoma have been reported in the postmarketing period for adolescent males with either ulcerative colitis or Crohn disease.
- Tuberculosis: Active tuberculosis or the reactivation of latent tuberculosis has been reported in patients receiving tumor necrosis factor-alpha inhibitors. Patients should be evaluated for the presence of tuberculosis before the initiation of therapy with tumor necrosis factor alpha inhibitors and treatment with antimycobacterial therapy should be initiated if latent tuberculosis is found. In cases of reactivation of latent tuberculosis, the reactivation occurs within the first few months of treatment with tumor necrosis factor-alpha inhibitors.
- Infection: Patients receiving treatment with a tumor necrosis factor-alpha inhibitor are at heightened risk of developing serious or fatal infections. This occurred more commonly in patients that are receiving treatment with other immunosuppressive agents including methotrexate and corticosteroids. The types of infections include fungal infections, such as aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis and pneumocystosis, and bacterial and viral infections. In particular, legionellosis and listeriosis have been reported. Patients should be monitored for signs of infection and treatment with tumor necrosis factor-alpha inhibitors should be discontinued if serious infection or sepsis occurs. Empiric antifungal therapy should be considered in patients who are at increased risk of invasive fungal infections.
- Salomon BL, Leclerc M, Tosello J, Ronin E, Piaggio E, Cohen JL. Tumor Necrosis Factor α and Regulatory T Cells in Oncoimmunology. Front Immunol. 2018;9:444. Published 2018 Mar 12. doi:10.3389/fimmu.2018.00444 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5857565
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin‐induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 1975; 72(9): 3666–3670.
- Pennica D, Hayflick JS, Bringman TS, Palladino MA, Goeddel DV. Cloning and expression in Escherichia coli of the cDNA for murine tumor necrosis factor. Proc Natl Acad Sci USA 1985; 82(18): 6060–6064.
- Robak T, Gladalska A, Stepien H. The tumour necrosis factor family of receptors/ligands in the serum of patients with rheumatoid arthritis. Eur Cytokine Netw 1998; 9(2): 145–154.
- Kwiatkowski D, Hill AV, Sambou I, Twumasi P, Castracane J, Manogue KR, et al. TNF concentration in fatal cerebral, non‐fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet 1990; 336(8725): 1201–1204.
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol (2003) 3:745–56.10.1038/nri1184
- Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor necrosis factor-alpha regulation of CD4+CD25+ T cell levels in NOD mice. Proc Natl Acad Sci U S A (2002) 99:12287–92.10.1073/pnas.172382999
- Mohan N, Edwards ET, Cupps TR, Oliverio PJ, Sandberg G, Crayton H, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum (2001) 44:2862–9.10.1002/1529-0131(200112)44:12<2862::AID-ART474>3.0.CO;2-W
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 2002 May 01;168(9):4620-7.
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour‐necrosis factor‐alpha from cells. Nature 1997; 385(6618): 729–733.
- Wang J, Al‐Lamki RS, Zhang H, Kirkiles‐Smith N, Gaeta ML, Thiru S, et al. Histamine antagonizes tumor necrosis factor (TNF) signalling by stimulating TNF receptor shedding from the cell surface and Golgi storage pool. J Biol Chem 2003; 278(24): 21751–21760.
- Chen G., Goeddel D.V. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924
- Locksley R.M., Killeen N., Lenardo M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/S0092-8674(01)00237-9
- Sun S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177
- Karin M., Delhase M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210
- Oltmanns U., Issa R., Sukkar M.B., John M., Chung K.F. Role of c-Jun N-terminal kinase in the induced release of GM-CSF, RANTES and Il-8 from human airway smooth muscle cells. Br. J. Pharmacol. 2003;139:1228–1234. doi: 10.1038/sj.bjp.0705345
- Festjens N., Vanden Berghe T., Cornelis S., Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell. Death Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085
- Riches D.W., Chan E.D., Winston B.W. TNF-alpha-induced regulation and signalling in macrophages. Immunobiology. 1996;195:477–490. doi: 10.1016/S0171-2985(96)80017-9
- Wajant H., Scheurich P. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in tnf signaling. Int. J. Biochem. Cell Biol. 2001;33:19–32. doi: 10.1016/S1357-2725(00)00064-9
- Bradley, J. (2008), TNF‐mediated inflammatory disease. J. Pathol., 214: 149-160. doi:10.1002/path.2287 https://doi.org/10.1002/path.2287
- Islam M.S., Ciavattini A., Petraglia F., Castellucci M., Ciarmela P. Extracellular matrix in uterine leiomyoma pathogenesis: A potential target for future therapeutics. Hum. Reprod. Update. 2018;24:59–85. doi: 10.1093/humupd/dmx032
- Wolanska M., Taudul E., Bankowska-Guszczyn E., Kinalski M. Tumor necrosis factor in uterine leiomyomas at various stages of tumor growth. Ginekol. Pol. 2010;81:431–434.
- Protic O., Toti P., Islam M.S., Occhini R., Giannubilo S.R., Catherino W.H., Cinti S., Petraglia F., Ciavattini A., Castellucci M., et al. Possible involvement of inflammatory/reparative processes in the development of uterine fibroids. Cell Tissue Res. 2016;364:415–427. doi: 10.1007/s00441-015-2324-3
- Postal M., Lapa A.T., Sinicato N.A., de Oliveira Pelicari K., Peres F.A., Costallat L.T., Fernandes P.T., Marini R., Appenzeller S. Depressive symptoms are associated with tumor necrosis factor alpha in systemic lupus erythematosus. J. Neuroinflamm. 2016;13:5. doi: 10.1186/s12974-015-0471-9
- Ciebiera M, Włodarczyk M, Zgliczyńska M, et al. The Role of Tumor Necrosis Factor α in the Biology of Uterine Fibroids and the Related Symptoms. Int J Mol Sci. 2018;19(12):3869. Published 2018 Dec 4. doi:10.3390/ijms19123869 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6321234
- Cerami A, Ikeda Y, Le Trang N, Hotez PJ, Beutler B. Weight loss associated with an endotoxin‐induced mediator from peritoneal macrophages: the role of cachectin (tumor necrosis factor). Immunol Lett 1985; 11(3–4): 173–177.
- Beutler BA, Milsark IW, Cerami A. Cachectin/tumor necrosis factor: production, distribution, and metabolic fate in vivo. J Immunol 1985; 135(6): 3972–3977.
- Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti‐cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987; 330(6149): 662–664.
- Fisher CJ Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, et al. Treatment of septic shock with the tumor necrosis factor receptor: Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med 1996; 334(26): 1697–1702.
- Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, et al. Mice deficient for the 55 kDa tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993; 73(3): 457–467.
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, et al. TNF receptor‐deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol 1998; 160(2): 943–952.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168(9): 4620–4627.
- Wallis RS, Broder M, Wong J, Lee A, Hoq L. Reactivation of latent granulomatous infections by infliximab. Clin Infect Dis 2005; 41(suppl 3): S194–198.
- Rudin W, Eugster HP, Bordmann G, Bonato J, Muller M, Yamage M, et al. Resistance to cerebral malaria in tumor necrosis factor‐alpha/beta‐deficient mice is associated with a reduction of intercellular adhesion molecule‐1 up‐regulation and T helper type 1 response. Am J Pathol 1997; 150(1): 257–266.
- Kwiatkowski D, Molyneux ME, Stephens S, Curtis N, Klein N, Pointaire P, et al. Anti‐TNF therapy inhibits fever in cerebral malaria. Qu J Med 1993; 86(2): 91–98.
- van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis 1996; 174(5): 1091–1097.
- Smith CA, Davis T, Anderson D, Solam L, Beckmann MP, Jerzy R, et al. A receptor for tumor necrosis factor defines an unusual family of cellular and viral proteins. Science 1990; 248(4958): 1019–1023.
- Creagan ET, Kovach JS, Moertel CG, Frytak S, Kvols LK. A phase I clinical trial of recombinant human tumor necrosis factor. Cancer 1988; 62(12): 2467–2471.
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice deficient in tumor necrosis factor‐alpha are resistant to skin carcinogenesis. Nat Med 1999; 5(7): 828–831.
- Grunhagen DJ, de Wilt JH, Graveland WJ, van Geel AN, Eggermont AM. The palliative value of tumor necrosis factor alpha‐based isolated limb perfusion in patients with metastatic sarcoma and melanoma. Cancer 2006; 106(1): 156–162.
- van Etten B, de Vries MR, van IMG, Lans TE, Guetens G, Ambagtsheer G, et al. Degree of tumour vascularity correlates with drug accumulation and tumour response upon TNFα‐based isolated hepatic perfusion. Br J Cancer 2003; 88(2): 314–319.
- Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, et al. Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J Immunol 1994; 152(8): 4149–4156.
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 1996; 14: 397–440.
- Williams RO, Feldmann M, Maini RN. Anti‐tumor necrosis factor ameliorates joint disease in murine collagen‐induced arthritis. Proc Natl Acad Sci USA 1992; 89(20): 9784–9788.
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU‐rich elements: implications for joint and gut‐associated immunopathologies. Immunity 1999; 10(3): 387–398.
- Elliott MJ, Maini RN, Feldmann M, Long‐Fox A, Charles P, Katsikis P, et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthrit Rheumat 1993; 36(12): 1681–1690.
- Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double‐blind randomized controlled trial. Lancet 2004; 363(9410): 675–681.
- Buch MH, Bingham SJ, Bryer D, Emery P. Long‐term infliximab treatment in rheumatoid arthritis: subsequent outcome of initial responders. Rheumatology 2007; 46(7): 1153–1156.
- Breese EJ, Michie CA, Nicholls SW, Murch SH, Williams CB, Domizio P, et al. Tumor necrosis factor alpha‐producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology 1994; 106(6): 1455–1466.
- Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak RN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350(9): 876–885.
- Sandborn WJ, Feagan BG, Hanauer SB, Present DH, Sutherland LR, Kamm MA, et al. An engineered human antibody to TNF (CDP571) for active Crohn’s disease: a randomized double‐blind placebo‐controlled trial. Gastroenterology 2001; 120(6): 1330–1338.
- Sandborn WJ, Feagan BG, Radford‐Smith G, Kovacs A, Enns R, Innes A, et al. CDP571, a humanized monoclonal antibody to tumour necrosis factor alpha, for moderate to severe Crohn’s disease: a randomized, double blind, placebo controlled trial. Gut 2004; 53(10): 1485–1493.
- Tumor necrosis factor-alpha antibody for induction of remission in Crohn’s disease. https://www.cochranelibrary.com/central/doi/10.1002/central/CN-01779632/full
- Tumour necrosis factor alpha blocking agents for induction of remission in ulcerative colitis. https://www.cochranelibrary.com/central/doi/10.1002/central/CN-01779637/full
- Braun J, Bollow M, Neure L, Seipelt E, Seyrekbasan F, Herbst H, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthrit Rheumat 1995; 38(4): 499–505.
- Francois RJ, Neure L, Sieper J, Braun J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann Rheumat Dis 2006; 65(6): 713–720.
- Lange U, Teichmann J, Stracke H. Correlation between plasma TNFα, IGF‐1, biochemical markers of bone metabolism, markers of inflammation/disease activity, and clinical manifestations in ankylosing spondylitis. Eur J Med Res 2000; 5(12): 507–511.
- Gorman JD, Sack KE, Davis JC Jr. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med 2002; 346(18): 1349–1356.
- Baraliakos X, Listing J, Rudwaleit M, Brandt J, Sieper J, Braun J. Radiographic progression in patients with ankylosing spondylitis after 2 years of treatment with the tumour necrosis factor alpha antibody infliximab. Ann Rheumat Dis 2005; 64(10): 1462–1466.
- Inman RD, Clegg DO, Davis JC, Whitmore JB, Solinger A. Etanercept in adult patients with early onset ankylosing spondylitis. J Rheumatol 2006; 33(8): 1634–1636.
- Baraliakos X, Listing J, Rudwaleit M, Brandt J, Alten R, Burmester G, et al. Safety and efficacy of readministration of infliximab after long‐term continuous therapy and withdrawal in patients with ankylosing spondylitis. J Rheumatol 2007; 34(3): 510–515.
- Ettehadi P, Greaves MW, Wallach D, Aderka D, Camp RD. Elevated tumour necrosis factor‐alpha (TNFα) biological activity in psoriatic skin lesions. Clin Exp Immunol 1994; 96(1): 146–151.
- Gottlieb AB, Evans R, Li S, Dooley LT, Guzzo CA, Baker D, et al. Infliximab induction therapy for patients with severe plaque‐type psoriasis: a randomized, double‐blind, placebo‐controlled trial. J Am Acad Dermatol 2004; 51(4): 534–542.
- Tyring S, Gordon KB, Poulin Y, Langley RG, Gottlieb AB, Dunn M, et al. Long‐term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol 2007; 143(6): 719–726.
- Feuerstein GZ, Liu T, Barone FC. Cytokines, inflammation, and brain injury: role of tumor necrosis factor‐alpha. Cerebrovasc Brain Metab Rev 1994; 6(4): 341–360.
- Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev 1999; 10(2): 119–130.
- Nawashiro H, Martin D, Hallenbeck JM. Neuroprotective effects of TNF binding protein in focal cerebral ischemia. Brain Res 1997; 778(2): 265–271.
- Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med 2001; 193(4): 427–434.
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001; 4(11): 1116–1122.
- TNF neutralization in MS: results of a randomized, placebo‐controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53(3): 457–465.
- Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor‐alpha‐induced cardiomyopathy. Circulation 2004; 109(15): 1892–1897.
- Suzuki J, Cole SE, Batirel S, Kosuge H, Shimizu K, Isobe M, et al. Tumor necrosis factor receptor‐1 and ‐2 double deficiency reduces graft arterial disease in murine cardiac allografts. Am J Transpl 2003; 3(8): 968–976.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti‐tumor necrosis factor therapy. Am J Med 2004; 116(5): 305–311.
- Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFα in pulmonary pathophysiology. Resp Res 2006; 7: 125.
- Cembrzynska‐Nowak M, Szklarz E, Inglot AD, Teodorczyk‐Injeyan JA. Elevated release of tumor necrosis factor‐alpha and interferon‐gamma by bronchoalveolar leukocytes from patients with bronchial asthma. Am Rev Resp Dis 1993; 147(2): 291–295.
- Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, et al. Tumour necrosis factor (TNFα) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 2005; 60(12): 1012–1018.
- Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med 2006; 354(7): 697–708.
- Al‐Lamki RS, Wang J, Vandenabeele P, Bradley JA, Thiru S, Luo D, et al. TNFR1‐ and TNFR2‐mediated signalling pathways in human kidney are cell type‐specific and differentially contribute to renal injury. FASEB J 2005; 19(12): 1637–1645.
- Vielhauer V, Stavrakis G, Mayadas TN. Renal cell‐expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J Clin Invest 2005; 115(5): 1199–1209.
- Lamprecht P, Voswinkel J, Lilienthal T, Nolle B, Heller M, Gross WL, et al. Effectiveness of TNFα blockade with infliximab in refractory Wegener’s granulomatosis. Rheumatology 2002; 41(11): 1303–1307.
- Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med 2005; 352(4): 351–361.
- Gartlehner G, Hansen RA, Jonas BL, Thieda P, Lohr KN. Biologics for the treatment of juvenile idiopathic arthritis: a systematic review and critical analysis of the evidence. Clin Rheumatol 2008; 27: 67–76.
- Baughman RP, Drent M, Kavuru M, Judson MA, Costabel U, du Bois R, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Resp Crit Care Med 2006; 174(7): 795–802.
- Efthimiou P. Tumor necrosis factor‐alpha in inflammatory myopathies: pathophysiology and therapeutic implications. Semin Arthrit Rheumat 2006; 36(3): 168–172.
- Ribi C, Sztajzel R, Delavelle J, Chizzolini C. Efficacy of TNFα blockade in cyclophosphamide‐resistant neuro‐Behcet disease. J Neurol Neurosurg Psychiat 2005; 76(12): 1733–1735.
- Sobrin L, Kim EC, Christen W, Papadaki T, Letko E, Foster CS. Infliximab therapy for the treatment of refractory ocular inflammatory disease. Arch Ophthalmol 2007; 125(7): 895–900.
- Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Therap 2002; 301(2): 418–426.
- Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995; 83(5): 793–802.
- Gottlieb AB. Tumor necrosis factor blockade: mechanism of action. J. Investig. Dermatol. Symp. Proc. 2007 May;12(1):1-4.
- Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, Weaver AL, Keystone EC, Furst DE, Mease PJ, Ruderman EM, Horwitz DA, Arkfeld DG, Garrison L, Burge DJ, Blosch CM, Lange ML, McDonnell ND, Weinblatt ME. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann. Intern. Med. 1999 Mar 16;130(6):478-86.
- Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, Smolen J, Emery P, Harriman G, Feldmann M, Lipsky P. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999 Dec 04;354(9194):1932-9.
- Mocci G, Marzo M, Papa A, Armuzzi A, Guidi L. Dermatological adverse reactions during anti-TNF treatments: focus on inflammatory bowel disease. J Crohns Colitis. 2013 Nov;7(10):769-79.





{kind=link}