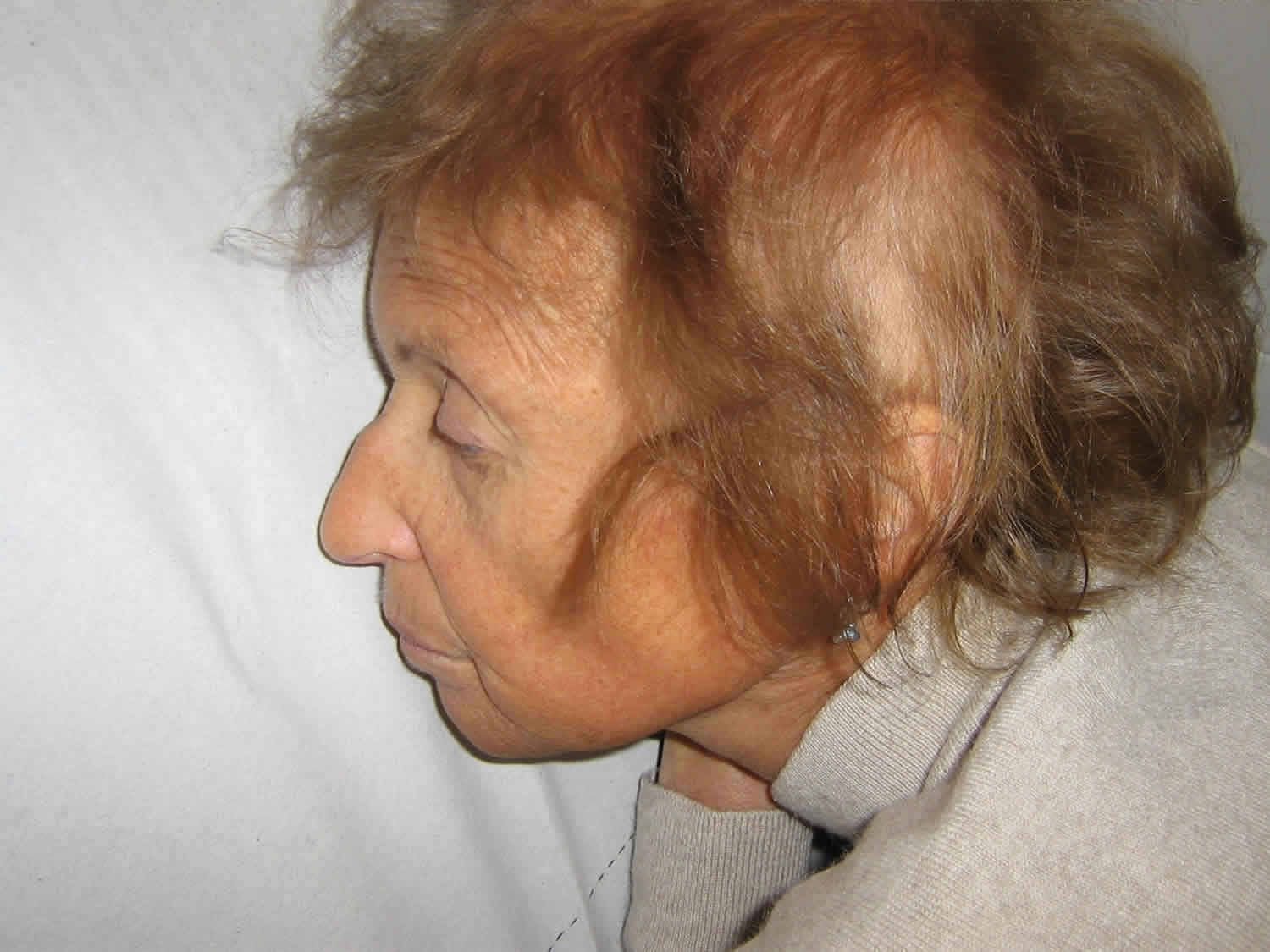
What is Werner syndrome
Werner syndrome is a rare inherited disorder that is characterized by the appearance of unusually accelerated aging (progeria). Although Werner syndrome is typically recognized by the third or fourth decades of life, certain characteristic findings are present beginning during adolescence and early adulthood. Individuals with Werner syndrome typically grow and develop normally until they reach puberty. Affected teenagers usually do not have a growth spurt, resulting in short stature and low weight relative to height. The characteristic aged appearance of individuals with Werner syndrome typically begins to develop when they are in their twenties and includes early graying (canities) and premature loss of scalp hair (alopecia). As Werner syndrome progresses, additional abnormalities include loss of the layer of fat beneath the skin (subcutaneous adipose tissue); a hoarse voice or a distinctive high-pitched voice; severe wasting (atrophy) of muscle tissue in certain areas of the body; and degenerative skin changes, particularly in the facial area, the upper arms and hands, and the lower legs and feet (distal extremities). Due to degenerative changes affecting the facial area, individuals with Werner syndrome may also have a facial appearance described as “bird-like” with unusually prominent eyes, a beaked or pinched nose, and/or other characteristic facial abnormalities. Many people with Werner syndrome have thin arms and legs and a thick trunk due to abnormal fat deposition.
As Werner syndrome progresses, affected individuals may develop disorders of aging early in life, such as premature clouding of the lenses of the eyes (bilateral senile cataracts) in both eyes, skin ulcers, certain endocrine defects, such as impaired functioning of the ovaries in females or testes in males (hypogonadism) with diminished fertility or abnormal production of the hormone insulin by the pancreas and resistance to the effects of insulin (type 2 diabetes) and some types of cancer. It is not uncommon for affected individuals to develop multiple, rare cancers during their lifetime. In addition, individuals with Werner syndrome may develop progressive thickening and loss of elasticity of artery walls (arteriosclerosis). Affected blood vessels typically include the arteries that transport oxygen-rich (oxygenated) blood to heart muscle (coronary arteries). Some affected individuals may also be susceptible to developing certain benign (noncancerous) or malignant tumors. Progressive arteriosclerosis, malignancies, and/or associated abnormalities may result in potentially life-threatening complications by approximately the fourth or fifth decade of life.
People with Werner syndrome usually live into their late forties or early fifties. The most common causes of death are cancer and atherosclerosis.
Werner syndrome is caused by abnormal changes (mutations) in the WRN gene. More than 80 different mutations of the WRN gene have been identified in individuals with the disorder. The WRN gene encodes for a “helicase” protein, suggesting that impaired DNA metabolism is involved in the premature aging seen in individuals with the disorder. Metabolism refers to the chemical processes occurring within bodily tissues. DNA or deoxyribonucleic acid, which is the carrier of the genetic code within cells, has a coiled (helical), ladder-like structure and is composed of strands of particular chemical groups. DNA “helicase” proteins are thought to promote the “unwinding” of DNA during certain cellular activities, such as the repair of damaged DNA and the separation of identical chromosomes (chromosomal segregation) into two “daughter cells” during cellular division and reproduction. Researchers suggest that Werner syndrome is due to complete loss of function of the helicase protein encoded by the WRN gene. The specific function of the helicase protein in preventing premature aging remains unclear.
Werner syndrome is inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits an abnormal gene from each parent. If an individual receives one normal gene and one abnormal gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the abnormal gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Werner syndrome is a rare disorder that affects males and females in equal numbers. Werner syndrome is estimated to affect 1 in 200,000 individuals in the United States 1. Werner syndrome occurs more often in Japan, affecting 1 in 20,000 to 1 in 40,000 people. Since the disorder was originally described in the medical literature in 1904, more than 800 cases have been reported.
Although certain associated findings are present beginning during childhood, puberty, and young adulthood, Werner syndrome is most frequently recognized in the third or fourth decades of life.
Figure 1. Werner syndrome
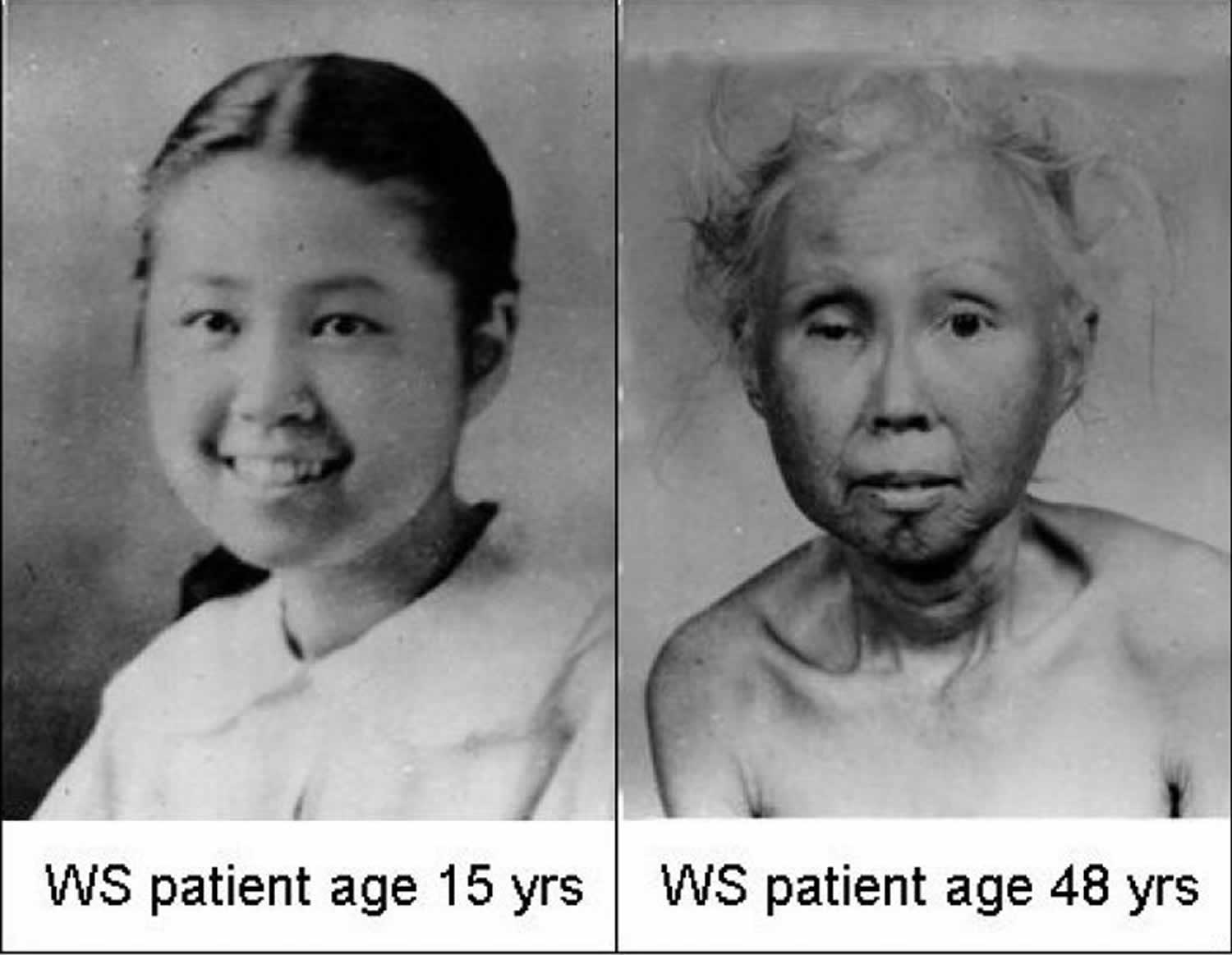
Footnote: The two photographs show a Japanese-American woman as a teenager and at 48 years of age.
[Source 2 ]Werner syndrome cause
Mutations in the WRN gene cause Werner syndrome. The WRN gene provides instructions for producing the Werner protein, which is thought to perform several tasks related to the maintenance and repair of DNA. This protein also assists in the process of copying (replicating) DNA in preparation for cell division. Mutations in the WRN gene often lead to the production of an abnormally short, nonfunctional Werner protein. Research suggests that this shortened protein is not transported to the cell’s nucleus, where it normally interacts with DNA. Evidence also suggests that the altered protein is broken down more quickly in the cell than the normal Werner protein. Researchers do not fully understand how WRN mutations cause the signs and symptoms of Werner syndrome. Cells with an altered Werner protein may divide more slowly or stop dividing earlier than normal, causing growth problems. Also, the altered protein may allow DNA damage to accumulate, which could impair normal cell activities and cause the health problems associated with this condition.
During laboratory (in vitro) studies of samples of skin cells (cultured human fibroblasts), researchers have demonstrated that the cells from individuals without the disorder may multiply approximately 60 times (“population doublings”) whereas Werner syndrome fibroblasts may reproduce only up to about 20 times. Due to such findings, some researchers have suggested that WRN is essentially a “counting gene,” regulating the total number of times that human cells may divide and reproduce. Such researchers speculate that mutations of the WRN gene may result in premature inhibition of DNA replication processes (synthesis) and early cellular aging (senescence), events that typically occur later in normal, aging human cells.
Researchers have also observed a high frequency of chromosomal abnormalities (e.g., random translocations) in cultured skin cells (fibroblasts) and cultured white blood cells (lymphocytes) derived from certain cell lines (clones) in individuals with Werner syndrome. Such findings (sometimes referred to as “variegated translocation mosaicism”) suggest that “chromosome breakage” may be characteristic of or play some role in the disease process. However, the specific implications of such findings remain unknown and further research is required.
Werner syndrome inheritance pattern
Werner syndrome is inherited in an autosomal recessive pattern, which means both copies of the WRN gene in each cell have mutations. The parents of an individual with Werner syndrome each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
The parents of some individuals with Werner syndrome have been closely related by blood (consanguineous). In these cases, if both parents carry the same disease gene, there is a higher-than-normal risk that their children may inherit the two disease genes necessary for the development of the disease.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
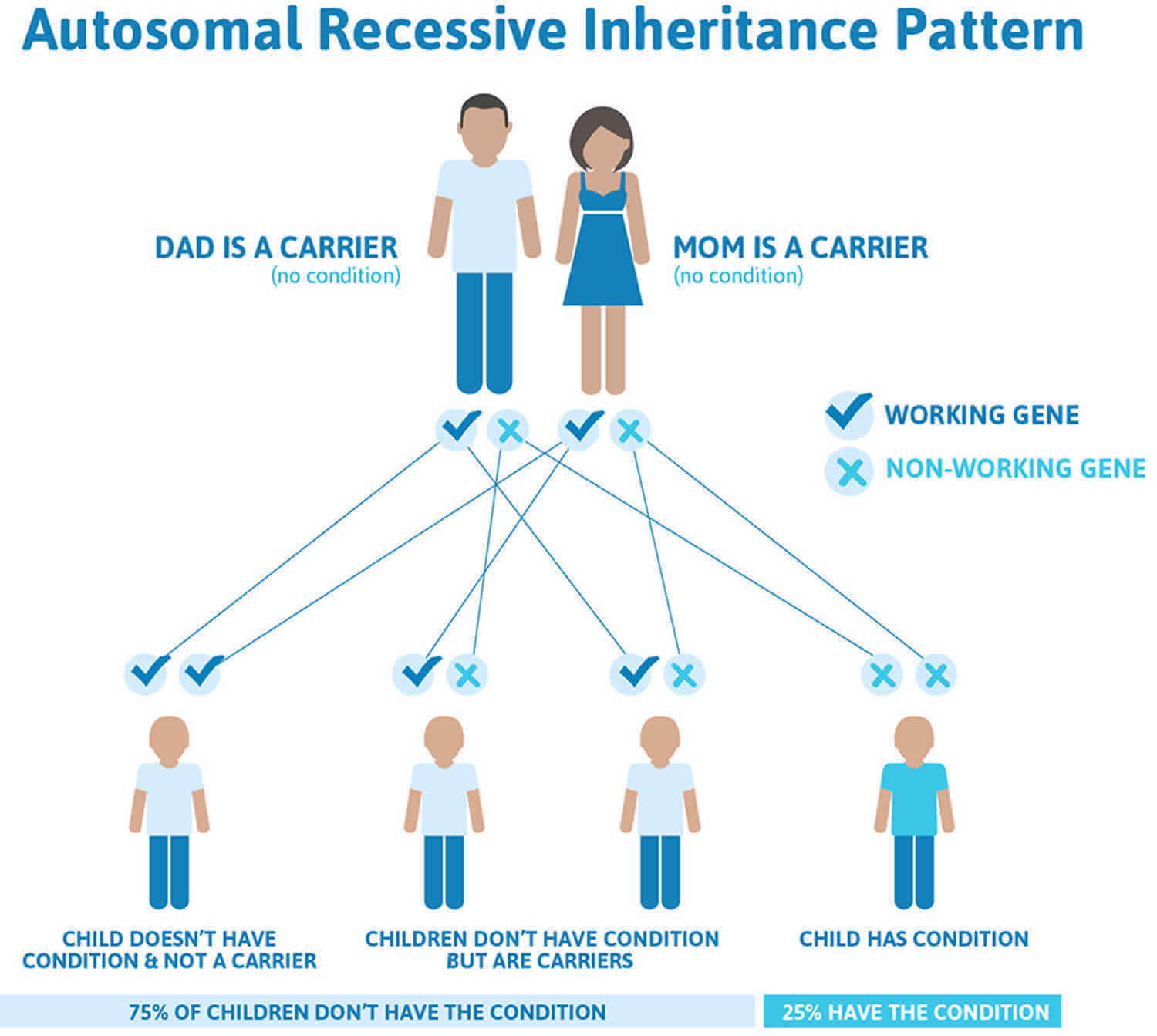
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Werner syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Werner syndrome symptoms
The signs and symptoms of Werner syndrome do not usually appear until the teens. This first sign may be slower than normal growth during puberty. In the 20s-30s, other signs of early aging appear. Some of the signs and symptoms of Werner syndrome include 3:
- Shorter than average height as an adult
- Bilateral cataracts (clouding of the lens of both eyes)
- Premature graying and thinning of the hair on the head
- Skin changes (thinning of skin, fragile skin)
- Loss of fat under the skin
- Thin limbs
- Characteristic facial changes (pinched nose, narrow face)
- Voice changes
- Decreased functioning of the testes and ovaries
- Decreased fertility
- Open skin sores, especially on the ankles
- Narrowing and hardening of the arteries (atherosclerosis)
- Type 2 diabetes mellitus
- Thinning of the bones (osteoporosis)
- Cancer
Not everyone with Werner syndrome will have all of these symptoms.
Children with Werner syndrome often appear unusually thin and, during late childhood, have an unusually slow growth rate. In addition, there is absence of the growth spurt typically seen during adolescence. Affected individuals typically reach their final height by approximately 13 years of age. However, adult height may be reached as early as at age 10 or as late as at age 18. Weight is also unusually low, even relative to short stature.
Before age 20, most individuals with Werner syndrome develop early graying and whitening of the scalp hair (canities). By about 25 years of age, affected individuals may experience premature loss of scalp hair (alopecia) as well as loss of the eyebrows and eyelashes. In addition, hair under the arms (axillary hair), in the pubic area, and on the trunk may be unusually sparse or absent. According to reports in the medical literature, the hair loss seen in those with Werner syndrome may occur secondary to impaired functioning of the ovaries in females or the testes in males (hypogonadism), an endocrine condition associated with deficient growth and sexual development. Both males and females with Werner syndrome may be affected by hypogonadism. As a result, affected males usually have an unusually small penis and small testes. Some females with the disorder may fail to develop secondary sexual characteristics (e.g., appearance of axillary and pubic hair, breast development, menstruation) and have poorly developed genitals. In other affected females, menstruation may be spare and irregular. Due to hypogonadism, most of those with the disorder may be infertile. However, there have been reports in the literature confirming that some affected males and females have reproduced.
In addition to premature graying and hair loss, individuals with Werner syndrome are affected by other progressive degenerative changes, including gradual loss of the layer of fat beneath the skin (subcutaneous adipose tissue); severe wasting (atrophy) of muscles within the hands, legs, and feet; and premature, generalized loss of bone density (osteoporosis), a condition that may cause or contribute to repeated fractures following minor trauma. Dental abnormalities may also be present, including abnormal development and premature loss of teeth. In approximately one third of individuals with Werner syndrome, there is also an abnormal accumulation of calcium salts (calcification) in and associated hardening of soft tissues (e.g., ligaments, tendons), particularly those of the elbows, knees, and ankles. In addition, due to progressive atrophy of the vocal cords, most individuals with the disorder develop an abnormally high-pitched voice. In other cases, the voice may be squeaky or unusually hoarse.
By approximately 25 years of age, individuals with Werner syndrome also develop progressive skin changes, particularly affecting the facial area, the upper arms and hands, and the lower legs and feet (distal extremities). For example, there is skin wasting (atrophy) over areas in which there is depletion of fatty (adipose), connective, and muscle tissue, resulting in the appearance of unusually shiny, “waxy,” smooth, or hardened (“scleroderma-like”) skin patches that may adhere to underlying tissues. Affected areas may be prone to developing open sores (ulcers) due to decreased supply of oxygenated blood to tissues (ischemia). The ulcers may be chronic and slow healing. Deep ulcerations around Achilles tendons and, less frequently, at elbows, are highly characteristic to Werner syndrome.
Many individuals with Werner syndrome also have additional skin abnormalities. Skin of the arms and legs may develop abnormally increased coloration (hyperpigmentation), decreased coloration (hypopigmentation), or abnormal widening of certain small underlying blood vessels, causing associated redness (telangiectasias). In addition, skin of the palms, of the soles, and overlying certain prominent joints, such as the elbows and knees, may become unusually thickened (hyperkeratosis) and tend to develop ulcers due to destruction of surface tissues.
Due to atrophic changes of the skin and underlying tissues in the facial area, affected individuals may have a distinctive, “pinched” facial appearance including unusually prominent eyes; stiff ears that have lost their elasticity; and a thin, beaked or pinched nose. Premature graying and loss of hair contribute to the characteristic appearance. According to reports in the medical literature, in most individuals with Werner syndrome, the appearance of premature aging is apparent by approximately age 30 to 40.
Werner syndrome is also typically characterized by the premature onset of senile cataracts, a condition in which there is loss of transparency of the lenses of the eyes. In individuals with Werner syndrome, cataracts typically affect both eyes (bilateral) and have an abrupt onset within the third or fourth decade of life. (Senile cataracts typically develop in individuals over age 50.) In some cases, other abnormalities of the eyes may also be present, such as an accumulation of calcium deposits within the transparent region in the front of the eyes (corneal calcification), inflammation of the middle and innermost layers of the eyes (chorioretinitis), degeneration of the nerve cells (rods and cones) of the retina that respond to light (retinitis pigmentosa), and/or progressive degeneration of the central region of the retina (senile macular degeneration). The degree of associated visual impairment depends upon the severity and/or combination of eye abnormalities present.
Approximately 70 percent of affected individuals have develop non-insulin-dependent (or type II) diabetes mellitus at the time of diagnosis. Non-insulin-dependent diabetes mellitus is a metabolic disorder characterized by resistance to the effects of the hormone insulin and abnormal insulin secretion by the pancreas, resulting in increased levels of the simple sugar glucose in the blood. (Insulin regulates glucose levels in the blood by promoting the movement of glucose into cells for energy production.) This form of diabetes usually develops in normal individuals of approximately 50 to 60 years. However, in those with Werner syndrome, the condition may become apparent by about age 35. Affected individuals may have no apparent symptoms (asymptomatic) at diagnosis or experience increased urination (polyuria), excessive thirst (polydipsia), increased hunger (polyphagia), and/or other characteristic symptoms. In addition, those with this form of diabetes may be susceptible to diabetic coma due to severely reduced levels of fluid within cells (hyperosmolar nonketotic coma). According to reports in the medical literature, although non-insulin-dependent diabetes mellitus may be associated with certain long-term complications, such as nerve damage (neuropathy), impaired kidney function (nephropathy), and damage to blood vessels within the retina (diabetic retinopathy), such complications have not been reported in affected individuals with Werner syndrome.
Werner syndrome is also characterized by severe, progressive, often widespread thickening and loss of elasticity of artery walls (arteriosclerosis). In some affected arteries, there may be abnormal accumulations of calcium deposits within the middle coat (tunica media) of the arteries and progressive destruction and replacement of the arteries’ muscle and elastic fibers with fibrous tissue (Monckeberg’s arteriosclerosis). Arteries affected by this form of arteriosclerosis may include those that transport oxygen-rich blood to heart muscle (coronary arteries) or certain arteries of the legs (peripheral vascular disease). Arteriosclerosis of peripheral blood vessels may cause or aggravate skin wasting (atrophy) and ulceration In addition, abnormal calcium deposits may accumulate within certain heart valves, such as the valve situated where the body’s major artery (aorta) arises from the lower left chamber of the heart (aortic valve) and the valve located between the left upper and lower heart chambers (mitral valve). Progressive arteriosclerosis may lead to episodes of chest pain due to deficient oxygen supply to heart muscle (anginal attacks); progressive inability of the heart to effectively pump blood to the lungs and the rest of the body (heart failure); localized loss of heart muscle caused by interruption of its blood supply (myocardial infarction or heart attack); and/or other potentially life-threatening complications.
People with Werner syndrome also have an increased predisposition to cancers. The most common neoplasms in Werner syndrome are carcinomas of thyroid, followed by cancers of the pigment-producing cells in skin and mucosa (malignant melanoma), cancer of the protective membranes surrounding the brain and the spinal cord (meningioma), tumors that arise within the soft tissues and bones (sarcomas and osteosarcoma), soft tissue sarcomas, primary bone tumors and leukemia/myelodysplasia.
Due to progressive arteriosclerosis, malignancies, and/or other associated abnormalities, many individuals with Werner syndrome may experience life-threatening complications by approximately the fourth or fifth decade of life.
Werner syndrome diagnosis
In some cases, Werner syndrome may be recognized clinically as early as approximately age 15, based upon a thorough clinical evaluation, characteristic physical findings (e.g., absence of growth spurt at puberty, short stature, low weight), and a careful patient and family history. However, the disorder often may not be recognized or confirmed until the third or fourth decades of life, once certain distinctive symptoms and findings are noted (e.g., premature graying and hair loss, distinctive voice, loss of subcutaneous tissue, muscular atrophy, skin changes, bilateral senile cataracts, etc.).
The diagnosis of Werner syndrome should be suspected in individuals who have the following cardinal signs 4:
- Bilateral ocular cataracts (present in 99%)*
- Premature graying and/or thinning of scalp hair (100%)
- Characteristic dermatologic pathology (96%)
- Short stature (95%)
Approximately 91% of affected individuals have all four cardinal signs.
The clinical diagnosis may be further supported by the presence of the following additional signs and symptoms 5:
- Thin limbs (present in 98%)
- Pinched facial features (96%)
- Osteoporosis (91%)
- Voice change (89%)
- Hypogonadism (80%)
- Type 2 diabetes mellitus (71%)
- Soft tissue calcification (67%)
- Neoplasm(s) (44%)
- Atherosclerosis (30%)
* Note: Percent frequencies are derived from individuals with a diagnosis of Werner syndrome confirmed by molecular testing.
Specialized imaging studies and laboratory tests may be conducted to detect, confirm, or characterize certain abnormalities potentially associated with the disorder. For example, eye specialists (ophthalmologists) may regularly monitor affected individuals for the development of cataracts with certain measures, such as use of a specialized instrument that enables visualization of the inside of the eyes (ophthalmoscope). If cataracts are detected, an illuminated microscope (slit lamp) may be used to examine the internal structures of the front regions of the eyes, enabling ophthalmologists to determine the specific location and extent of the cataracts.
Diagnostic testing may include monitoring of blood sugar levels to ensure prompt detection of diabetes mellitus, bone scans and blood tests for osteoporosis, and/or other studies. In addition, thorough cardiac evaluations and ongoing monitoring may also be performed (e.g., clinical examinations, X-ray studies, specialized cardiac tests) to assess associated cardiovascular abnormalities and determine appropriate disease management. Individuals with Werner syndrome should also be regularly monitored as necessary to ensure the prompt detection and appropriate treatment of certain malignancies or benign tumors that may occur in association with the disorder (e.g., osteosarcoma, meningioma).
In some cases, specialized laboratory tests may be performed on cultured skin cells (fibroblasts) from affected individuals, demonstrating abnormally decreased replication of Werner syndrome fibroblasts. Evaluation of the chromosomal make-up (karyotype) within the nuclei of cultured fibroblasts and certain white blood cells (lymphocytes) may reveal a high frequency of certain chromosomal rearrangements (variegated translocation mosaicism). (For more information, please see the “Causes” section of this report above.) In addition, according to several investigators, urine tests may reveal elevated levels of hyaluronic acid, a complex carbohydrate that is present in the spaces between certain cells (intercellular spaces) within certain connective tissues. The implications of this finding are not understood.
Confirmation of a clinical diagnosis of Werner syndrome may be achieved through molecular testing of the WRN gene. Molecular sequencing of the WRN gene to detect disease-causing mutations, as well as biochemical testing to quantitate the amount of WRN protein produced by cells, is available on a clinical basis.
Werner syndrome treatment
The treatment of Werner syndrome is directed toward the specific symptoms that are apparent in each individual. Disorder management may require the coordinated efforts of a team of specialists who may need to systematically and comprehensively plan an affected individual’s treatment. Such specialists may include internists; physicians who diagnose and treat disorders of the skeleton, muscles, joints, and other related tissues (orthopedists); physicians who diagnose and treat abnormalities of the heart and its major blood vessels; eye specialists (ophthalmologists); physicians who diagnose and treat disorders of the endocrine system (endocrinologists); and/or other health care professionals.
Specific therapies for individuals with Werner syndrome are symptomatic and supportive. According to reports in the medical literature, diabetes mellitus is typically mild and may often be managed with dietary changes and appropriate medications by mouth to decrease elevated sugar (glucose) levels in the blood (oral hypoglycemic medications).
In affected individuals with cataracts, treatment may include surgical removal of the clouded lens and implantation of a substitute lens (intraocular lens) or prescription of corrective glasses or contact lenses. Some physicians report that individuals with Werner syndrome may have a significantly increased risk of separation of surgical wound layers (wound dehiscence) and/or other complications (e.g., corneal endothelial decompensation). Therefore, these physicians recommend that special precautions be taken during such surgical procedures (e.g., small surgical incisions, avoidance of local or systemic cortisone).
In individuals with Werner syndrome, measures to manage arteriosclerosis and associated cardiovascular abnormalities are symptomatic and supportive. For example, in those with episodes of chest pain due to deficient oxygen supply to heart muscle (anginal attacks), treatment may include the use of certain medications that may help to minimize or manage such symptoms.
If benign or malignant tumors develop in association with Werner syndrome, appropriate treatment measures may vary depending upon the specific tumor type present; whether the tumor is benign or malignant; stage, grade, and/or extent of disease; and/or other factors. Depending upon such factors, treatment methods may include surgery, use of certain anticancer drugs (chemotherapy), radiation therapy, and/or other measures.
Genetic counseling is recommended for individuals with Werner syndrome and their families.
Werner syndrome life expectancy
Werner syndrome prognosis is unfavorable. The mean survival for patients with Werner syndrome is 46 years. Death usually occurs when patients are aged 30-50 years because of atherosclerosis or malignant tumors. Adroit medical management may enhance life expectancy; one patient was described who survived until dying of acute heart failure at age 68 years 6.
References- Werner syndrome. https://ghr.nlm.nih.gov/condition/werner-syndrome
- Salk, D. (1982). Werner’s syndrome: A review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Human Genetics, 62, 1-15.
- Oshima J, Sidorva JM, Monnat RJ Jr. Werner syndrome: Clinical features, pathogenesis, and potential therapeutic interventions. Ageing Res Rev. Jan 2017; 33:105-114. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5025328
- Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, Lucci-Cordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, von Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27:558–67.
- Oshima J, Martin GM, Hisama FM. Werner Syndrome. 2002 Dec 2 [Updated 2016 Sep 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1514
- Kawai T, Nozato Y, Kamide K, Onishi M, Yamamoto-Hanasaki H, Tatara Y, et al. Case report of a long-surviving Werner syndrome patient with severe aortic valve stenosis. Geriatr Gerontol Int. 2012 Jan. 12(1):174-5.





{kind=link}