
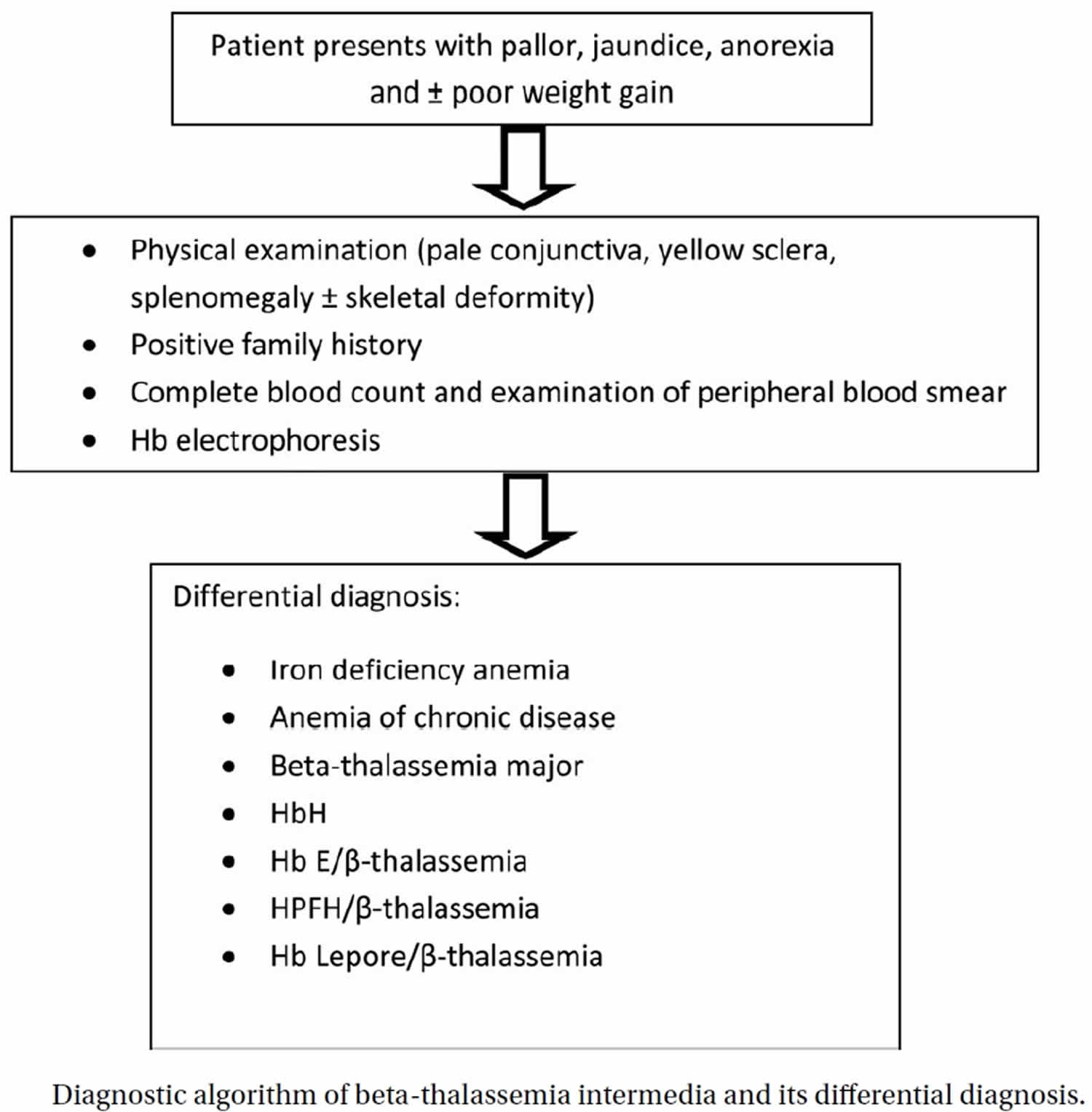
Thalassemia intermedia
Thalassemia intermedia describes a form of thalassemia of intermediate severity between the major, transfusion-dependent forms of the disease and the symptomless carrier states 1. The term thalassemia intermedia includes many different varieties of thalassemia including the compound heterozygous state for mild and severe beta thalassemia mutations or forms of homozygous beta thalassemia in which genetic modifiers have reduced the severity of the disease, diseases like hemoglobin E beta thalassemia due to the co-inheritance of a structural hemoglobin variant with beta thalassemia, the compound heterozygous states for other structural variants such as hemoglobins S or C and beta thalassemia and a heterogeneous group of forms of alpha thalassemia that produce hemoglobin H disease of varying severity 1. The wide clinical variability of thalassemia intermedia leads to major difficulties in their management. These problems have been magnified over recent years by the discovery that in many forms of thalassemia intermedia there are a wide range of complications which tend to occur later in the lives of affected patients.
Thalassemias are a group of inherited autosomal recessive blood disorders that affect the way the body makes hemoglobin (Hb). Hemoglobin (Hb) is a protein found in red blood cells that carries oxygen throughout the body. Hemoglobin is made up of alpha globin and beta globin. Differentiation between thalassemia major and thalassemia intermedia at presentation is not uniformly characterized, for which an absolute criteria needs to be developed 2.
The body contains more red blood cells than any other type of cell, and each has a life span of about 4 months. Each day, the body produces new red blood cells to replace those that die or are lost from the body.
With thalassemia, the red blood cells are destroyed at a faster rate, leading to anemia, a condition that can cause fatigue and other complications.
Thalassemias are inherited conditions — they’re carried in the genes and passed on from parents to children. People who are carriers of a thalassemia gene show no thalassemia symptoms and might not know they’re carriers. If both parents are carriers, they can pass the disease to their kids. Thalassemias are not contagious.
While there are many different types of thalassemias, the main two are:
- Alpha thalassemia: when the body has a problem producing alpha globin
- Beta thalassemia: when the body has a problem producing beta globin
When the gene that controls the production of either of these proteins is missing or mutated, it results in that type of thalassemia.
Beta thalassemia intermedia
The term beta thalassemia intermedia is clinically descriptive of beta-thalassemic patients whose clinical manifestations are not as mild as thalassemia minor or as severe as beta thalassemia major 3. The first description of beta thalassemia intermedia wasmade by Rietti GreppiMicheli in 1955 4. He described a thalassemic patient with clinical phenotype between thalassemia minor and beta thalassemia major 5. Beta thalassemia intermedia is when both beta globin genes are mutated, but the mutations are less severe. Thalassemia intermedia arises from defective gene function leading to partial suppression of beta-globin protein production. Thalassemia intermedia usually results from a homozygous or a compound heterozygous mutation 6. The reason for thalassemia intermedia is caused by 3 different mechanisms. The first is the inheritance of a mild or silent beta chain mutation, which keeps a low level of beta chains, as opposed to its absence in more severe cases making less of an alpha/beta imbalance. The second is the inheritance of determinants associated with increased gamma chain production, which pair with unbound alpha chains. The third is the co-inheritance of alpha-thalassemia, which decreases the number of unpaired chains due to decreased alpha chain synthesis 7. The genotypes of beta thalassemia intermedia are much complicated referring to β+/β+,β+/β0, Hb E/β0, β0/β0 compounding alpha thalassemia and so on 8. Beta thalassemia intermedia is neither as severe as beta thalassemia major nor as mild as beta thalassemia trait (beta thalassemia minor) 9.
Genetic heterogeneity of beta thalassemia intermedia is associated with wide clinical spectrum presentations from mild to severe hemolytic anemia and can be divided into two subgroups:
- Some patients are mildly affected leading to mild clinical problems until adult life. These patients maintain hemoglobin levels between 7 and 11 gr/dL and are usually transfusion independent or rarely require blood transfusions.
- Patients with more severe anemia who generally present at ages 2–6 years old. Although they may not require regular transfusions like the first subgroup, without occasional transfusions and appropriate management, they frequently develop clinical symptoms such as skeletal deformities and growth retardation 10.
While beta thalassemia intermedia and beta thalassemia major have some overlap in their clinical presentations, differentiation of the two disorders is essential for optimal management
and prevention of their later complications. Beta thalassemia intermedia can present with pallor, jaundice, anemia, splenomegaly or skeletal deformities during childhood or later. Diagnosis of beta thalassemia intermedia is usually made after the age of 2 years with initial Hb levels of 7 gr/dL or more in patients with beta-thalassemia who are free of infection and have adequate folic acid 11. One of their parents is also atypical carrier of beta-thalassemia such as normal or borderline HbA2 or isolated increased HbF (usually up to 10%) 10. The patients are usually referred with microcytic-hypochromic anemia (low MCV and low MCH) and the peripheral smear shows mild to severe microcytosis and hypochromia, anisopoikilocytosis, polychromasia, target cell, basophilic stippling, and nucleated red blood cell. Hb electrophoresis includes: HbA: up to 80%; HbA2: normal or up to 7%; HbF: > 10%. Serum iron, serum ferritin and transferrin saturation may be increased. Diagnostic algorithm of beta thalassemia intermedia and its differential diagnosis is shown in Figure 1. Differential diagnosis between beta thalassemia intermedia and beta thalassemia major is essential because the first step for management of patients with beta thalassemia intermedia is usually not transfusion; however, the first choice ofbeta thalassemia majormanagement is blood transfusion. Table 1 shows some differentiating parameters between beta thalassemia intermedia and beta thalassemia major.
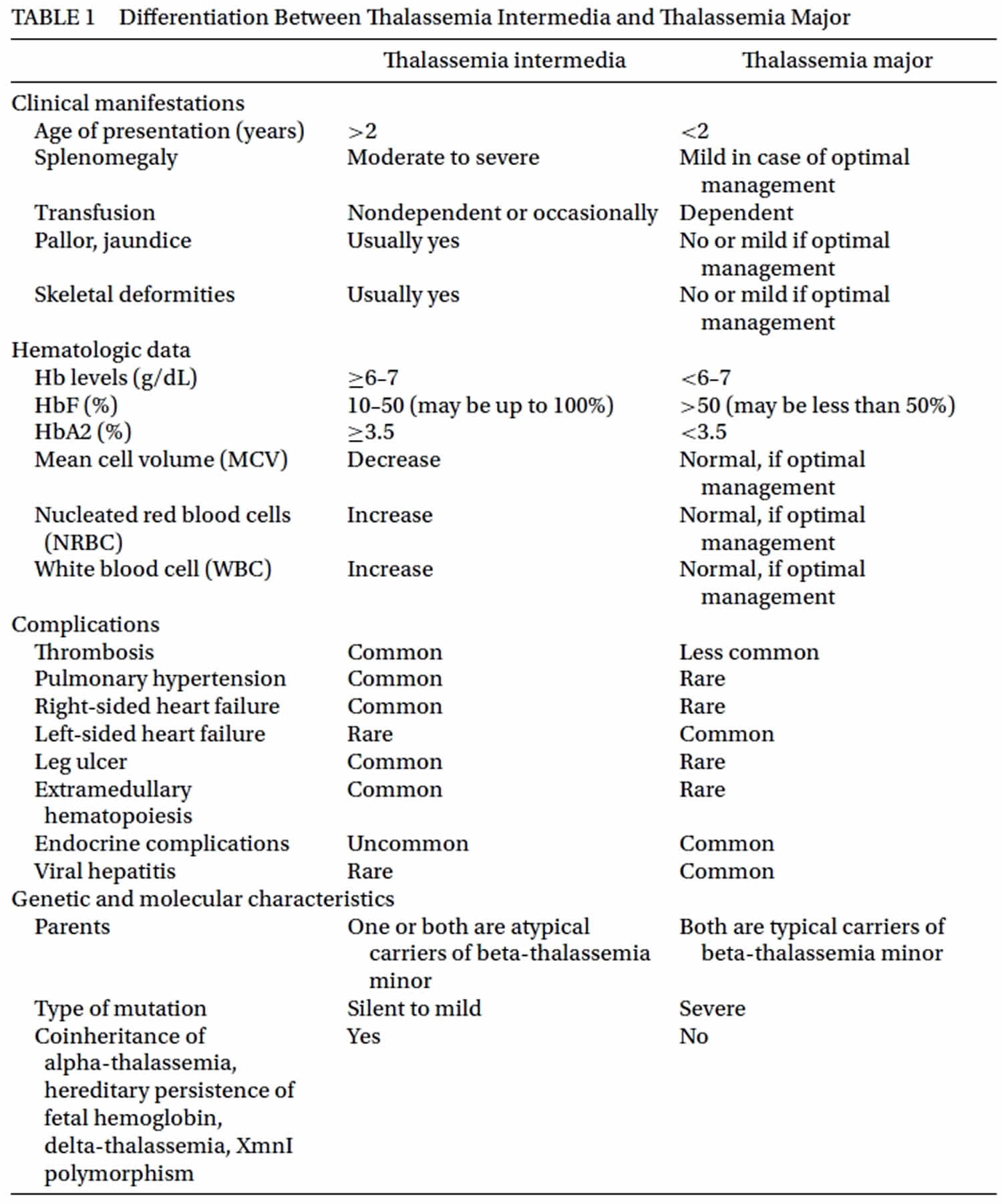
Figure 1. Beta thalassemia intermedia diagnostic algorithm
Beta thalassemia intermedia symptoms
The clinical manisfestation of beta thalassemia intermedia patients are heterogeneous. Some thalassemia intermedia patients are asymptomatic until adult life, whereas others are symptomatic from as young as 2 years of age with anemia and usually require infrequent blood transfusion after the first 2 years of life 12. Many patients with thalassemia intermedia receive only occasional or no transfusions, since they are able to maintain hemoglobin levels between 7-9 g/dL 13. Approximately 5%‐10% of thalassemia intermedia patients could live without requiring any blood transfusion 14. Individuals with beta thalassemia intermedia are at risk for iron overload secondary to increased intestinal absorption of iron as a result of ineffective erythropoiesis. Ineffective erythropoiesis is responsible for erythroid marrow hyperplasia and skeletal
deformities, hemolytic anemia and extramedullary hematopoiesis. Hemolysis is commonly associated with splenomegaly, hypercoagulable state, and pulmonary hypertension 11.
Hemolytic anemia
About 49.8% of beta thalassemia intermedia patients have always been transfusion independent, while 50.2% have been on transfusion dependent but still less than six packs per year in accordance with the definition 2. Kaddah N et al 15 reported 51.7% had no history of blood transfusion while 48.3% had history of blood transfusion. This is in agreement with Taher et al 16 in which 52% received no blood transfusions, while 48% received infrequent blood transfusions.
Mean hemoglobin was 7.77 g/dL in transfusion independent and 7.4 g/dL in transfusion dependant intermedia patients 2. Mean HbF of 93% with genetic characteristics of beta thalassemia is a confirmatory test for thalassemia syndrome. This finding was in contrast to the results reported by Kaddah N et al 15 with HbF ranged between 3.1 and 98 with a mean 26.47% and Qatanani et al 17 in which Hb F mean was 21.04%.
Mean volume of packed red cell transfused per year was 821.3 mL in thalassemia intermedia, which is justified by the results reported by Taher et al 18 as patients with thalassemia intermedia may benefit from an individually tailored transfusion regimen, compared with the regular transfusion regimens implemented in beta thalassemia major, to help prevent transfusion dependency. 2412.5 mL was the average packed red cell volume transfused per year in beta thalassemia major patients. Liver and spleen size was enlarged but not massively. Mean serum ferritin of 368 ng/mL and 1496.19 ng/mL showed iron overload in intermedia patients and major patients, respectively. In patients with non-transfusion dependent thalassemia, intestinal absorption of iron can be as much as 3‐4 mg/day or 1000 mg/year 19.
Extramedullary hematopoiesis
Erythropoietic tissue masses occur as a compensatory mechanism to overcome chronic hemolysis, erythroid marrow hyperplasia is most commonly found in the liver, spleen, and lymph nodes. Paraspinal masses can cause cord compression and neurological damage. Paraspinal erythroid marrow hyperplasia mainly presents as pseudo-tumors, which may possibly cause a variety of neurological symptoms due to spinal compression. Most of the cases remain asymptomatic, but various neurological clinical presentations have been reported.
Thrombosis
In a cohort study that was done on 8860 thalassemia patients (6670 beta thalassemia major and 2190 beta thalassemia intermedia) it was demonstrated that thromboembolic event occurred 4.38 times more frequently in beta thalassemia intermedia than beta thalassemia major patients 20. The most important factor involved in hypercoagulability is exposure of negatively charged phospholipids on the red blood cells membrane remnants as a result of oxidative stress and subsequent activation of the prothrombinase complex and enhanced thrombin generation. However, some other factors like decreased levels of the antithrombotic proteins C and S, endothelial exposure to inflammation, and oxidative effects of hemolysis and increased number of activated platelets in splenectomized patients are also involved in thromboembolic events in these patients. Splenectomized beta thalassemia intermedia patients have a higher incidence of thromboembolic event compared to non-splenectomized beta thalassemia intermedia patients 21. These patients who develop thromboembolic event are characterized by high nucleated red blood cell and platelet counts, and are more likely to have evidence of pulmonary hypertension and be transfusion naive. Furthermore, high nucleated red blood cell and platelet counts as well as transfusion naivety are associated with earlier development of thromboembolic event after splenectomy 22.
Deep and portal vein thrombosis, pulmonary embolism, and brain ischemia and infarction resulting in stroke are the main thromboembolic complications in beta thalassemia intermedia patients 23. Overt stroke is much more common in beta thalassemia majorthan beta thalassemia intermedia due to higher stroke related risk factors such as diabetic mellitus, heart failure and arrhythmia in these patients but silent ischemic brain lesions are more common in beta thalassemia intermedia. The initial study of neurologic disease in beta thalassemia intermedia from 1999 assessed brain MRI in 16 patients and
showed silent ischemic brain lesion in 37.5% of patients 24. Similar studies in Iran and Lebanon also showed the frequency of silent ischemic brain lesions in 26% and 60% of these patients, respectively 25.
Cerebrovascular accidents and silent ischemic lesions are reported especially frequently in beta thalassemia intermedia patients in the subgroup of patients who are adults, transfusion independent, splenectomized, and have a platelet count>500×10 9/L. It is highly recommended that patients with silent infarcts be treated with antiplatelet drugs. Blood transfusion on a regular basis should be strongly considered in such patients and definitely initiated for patients with symptomatic central nervous system disease 26. There is also a large study in Iran of 95 beta thalassemia intermedia patients that showed the protective effect of hydroxyurea therapy in the prevention of silent ischemic brain lesions 27. Thrombocytosis is a risk factor for thrombotic events, especially in splenectomized patients. Antiplatelet drugs like aspirin are indicated in this situation. Prophylactic anticoagulation therapy is also recommended in beta thalassemia intermedia patients who are undergoing some types of surgery as well as those who have a previous history of deep vein thrombosis, pulmonary embolism or stroke. Low molecular weight heparin can be used for a period of 7–14 days postoperatively to prevent postsurgery thrombosis. However, in patients with thromboembolic event life-long anticoagulation seems to be rational and effective in prevention of recurrent thromboembolic event.
Pulmonary hypertension
Pulmonary hypertension, defined as systolic pulmonary artery pressure >35 mmHg, may be a common complication in beta thalassemia intermedia patients with a frequency that has been reported to be as high as 60%. However, other studies found a frequency of 10–12% 28. Pulmonary hypertension is the primary cause of right sided congestive heart failure in these patients, in contrast to beta thalassemia major patients in whom left-sided ventricular failure is more common. Hemolysis has a key role in the development of pulmonary hypertension in beta thalassemia intermedia patients. It was shown that chronic hemolysis leads to nitric oxide depletion due to nitric oxide scavenging, arginine catabolism, and endogenous nitric oxide synthesis inhibition. It also contributes to enhanced platelet activation and increased endothelin-1 release 29, and thus, endothelial dysfunction, increased vascular tone, inflammation, hypercoagulability, vascular remodeling, and destruction of pulmonary vasculature, which ultimately results in hemolytic
anemia–associated pulmonary hypertension 30.
Blood transfusion and sildenafil (in some cases) are recommended as an optimal therapy in β-beta thalassemia intermedia patients with pulmonary arteria hypertension. Some studies have shown that hydroxyurea therapy alone or in combination with l-carnitine ormagnesium can be effective in improving hematologic parameters and cardiac status in patients with beta thalassemia intermedia 31.
Iron overload and Endocrine complications
Increased iron absorption due to chronic hemolytic anemia can cause iron overload and serious iron-related organ complications like cardiac dysfunction and endocrine dysfunction including diabetic mellitus, hypogonadism, infertility, and hypoparathyroidism 32. Endocrine complications are less common in beta thalassemia intermedia patients in comparison with beta thalassemia major patients because of fewer blood transfusions and therefore less severe overload.
Viral infection commonly due to blood borne viruses such as hepatitis C is a significant cause of liver disease in beta thalassemia intermedia.
Treatment of hepatitis C with α-interferon and ribavirin in these patients is associated with difficulty due to hemolysis from treatment which has exacerbated anemia and blood transfusionmay be needed 33.
Involvement of liver by iron overload or hepatitis or both can lead to cirrhosis and hepatocellular carcinoma in older patients 34.
Leg ulcer
Leg ulcer is a serious complication in beta thalassemia intermedia patients occurring in almost one third of patients with poorly controlled disease. They usually appear in the second decade of life and are generally located on the medial or lateral malleoli. The ulcers can develop afterminor trauma and tend to expand rapidly. Chronic anemia, reduced oxygen delivery to the distal regions and venous stasis in beta thalassemia intermedia patientsmay cause leg ulcers in older patients. Also, increased rigidity of erythrocytes cellular membrane, local edema due to venous stasis, right sided heart failure, repetitive local trauma, skin infections, hypercoagulablility, and prothrombotic tendency are other contributing factors to ulcer formation. Blood transfusion (in some cases), local wound care, and hydroxyurea therapy are the major strategies in these cases. Although hydroxyurea therapy specifically seems to help in beta thalassemia intermedia, it does not help in leg ulcers in sickle cell disease; however, it is associated with ulcer development in other conditions 35.
Other complications
Cachexia and hyperuricemia due to the hypercatabolism of erythroid hyperplastic tissue (more prevalent in beta thalassemia intermedia in comparison to beta thalassemia major) and cholelithiasis induced by hyperbillirubinemia are some other metabolic complications in beta thalassemia intermedia 36. Alloporinol therapy is recommended in cases with hyperuricemia.
Beta thalassemia intermedia treatment
Treatment of beta thalassemia intermedia is essential for prevention of clinical complications. The limited options available for management of beta thalassemia intermedia include transfusion therapy, iron chelation, splenectomy, modulation of gamma-globulin chain production, and stem cell transplantation. The OPTIMAL CARE study, performed to evaluate the rate of complications
in relation to currently practiced treatment options 21 showed that the highest rates of complications were seen in patients who did not receive any treatment (Table 2). This study showed the incidence of complications was lower among patients who received hydroxyurea therapy, transfusion and iron chelation therapy compared to those who
did not.
The size and location of erythropoietic tissue lesions as well as the extent of spinal cord involvement determine the severity, andmultiplicity of signs and symptoms 37. Erythroid marrow hyperplasia has also been reported in pleura, pericardium, chest, intracranial cavity, adrenal glands, and some other organs 38. Hydroxyurea therapy, blood transfusion, and radiation therapy are therapeutic options for erythroid marrow hyperplasia in these patients. There are several reports showing that hydroxyurea can be effective and safe. The dosage of hydroxyurea should be higher (20–30 mg/kg/day) compared to the dosage that it is usually used for beta thalassemia intermedia for enhancement of gamma globin chain synthesis (8–15 mg/kg/day).There are four case reports (3 beta thalassemia intermedia and 1 beta thalassemia major) that were successfully treated with hydroxyurea alone 39. The other treatment modalities should be considered if the patient does not respond to hydroxyurea therapy. Surgery is not recommended as it has been related to bleeding in these patients.

Transfusion therapy
Administration of regular blood transfusions is not a routine treatment strategy in all beta thalassemia intermedia patients but it is an essential treatment option in some situations (Table 3). Recently published international guidelines for transfusion therapy are numerous. Occasional transfusion should be done in pregnancy, surgery, and infections. More frequent transfusions should be considered in patientswith a declining hemoglobin level in parallel with profound enlargement of the spleen, patientswith growth failure, poor performance at school, diminished exercise tolerance, and failure of secondary sexual development in correlation with bone age. Furthermore, signs of bone changes, and poor quality of life should prompt initiation of blood transfusion therapy. Transfusion may also be considered for the primary prevention, management or secondary prevention of patients having thrombotic or cerebrovascular disease, patients with pulmonary hypertension with or without secondary heart failure, erythroid marrow hyperplasia, and leg ulcers. Alloimmunization to red cell antigens in beta thalassemia intermedia patients is more common than in beta thalassemia major and may be related to the later age atwhich transfusions are begun. Alloimmunization is also more common in splenectomized patients 40. Pretransfusion red cell antigenmatching, particularly for the Rh and Kell systems, is recommended.

Iron chelation therapy
Removal of excessive iron is essential inbeta thalassemia intermedia patientswith iron overload. Iron overload in beta thalassemia intermedia is frequently derived from transfusion therapy. In addition, chronic hemolysis, ineffective erythropoiesis, and hypoxiamay lead to increased intestinal iron absorption through suppression of the regulatory protein hepcidin 41. Until recently, the cutoff for starting chelation was an liver iron concentration of 7 and above. A recent study, however, showed that complications were more likely to occur at an liver iron concentration of 7 Fe/g dry weight and above, thus the need to start chelating patients earlier 42. Data from the first and largest randomized clinical trial of the iron chelator deferasirox in 166 patients (THALASSA) 41 showed that deferasirox therapy causes a significant reduction in liver iron concentration compared to patients on placebo, following 1 year therapy in patients above the age of 10 and with a baseline iron concentration of more than 5mg Fe/g dry weight. The Thalassemia International Federation recommends to initiate iron chelation therapy corresponding to a ferritin level of above 800 ng/mL and an liver iron concentration of 5mg Fe/g dry weight or above. Suspension of therapy should be initiated when serum ferritin level is 300ng/ml corresponding to an liver iron concentration level of 3 mg Fe/g dry weight or less. The Thalassemia International Federation also recommends that all patients above the age of 10 be frequently evaluated for iron overload by liver iron concentration at 1–2 year intervals along with serial measurements of serum ferritin every 3 months.
It can be said that serum ferritin alone is not a reliable measure of iron overload in these patients because it underestimates the iron load, and annual assessment of liver iron concentration by biopsy or preferably by noninvasive imaging methods like R2 and T2∗ MRI sequences havemore reliable and reproducible results 41. There are few clinical studies of chelation therapy in beta thalassemia intermedia patients, but all three licensed chelators including deferoxamine (Desferal), deferiprone (Ferriprox), and deferasirox (Exjade) achieve a good response 43. Compliance may be better with deferasirox than with deferoxamine or deferiprone 44 because it requires only one oral dose daily. Nevertheless, renal function should be closely evaluated due to more hyperuricemia and hypercatabulism in these patients compared to beta thalassemia major patients. In one study, hyperurecemia andmicroscopic hematuria were found to be more common in beta thalassemia intermedia than beta thalassemia major 45.
Modulation of gamma-globin chain production
Enhanced gamma-globin chain production might address some of the major clinical manifestations in beta thalassemia intermedia patients by increasing the production of HbF and reducing
the alpha/beta-globin chain imbalance. One of the best known drugs to enhance gamma-globin chain production is hydroxyurea. The clinical impact of hydroxyurea in treating beta thalassemia intermedia has been well documented in a few recent studies. The results show that a significant number of transfusion-dependent beta thalassemia intermedia patients became transfusion free or needed only occasional transfusions. Significant increases of Hb levels in not transfused patients were also reported 46. A recent study showed that combination therapy of hydroxyurea
(8–15mg/kg/d)with L-carnitine ormagnesium chloride could bemore effective in improving hematologic parameters and cardiac status in patients with beta thalassemia intermedia than hydroxyurea alone 47. Also, it has been reported thathydroxyurea therapymay be associated with decreased risk of stroke 27.
Hydroxyurea may lower the risk of thrombotic events by reducing phospholipid expression on the surface of erythrocytes and platelets and thereby reduce thrombin generation 3. Moreover, the OPTIMAL care study (the largest overview on the current status of beta thalassemia intermedia patients in 6 comprehensive care centers) demonstrated that hydroxyurea can be protective against osteoporosis, pulmonary hypertension, erythroid marrow hyperplasia, and thrombosis 21. Co-inheritance of alpha thalassemia, Hb E/β thalassemia, homozygosity for certain β-globin mutations [IVS II-I (G>A) or IVS I-V(G>C)], younger age, higher Hb levels, higher age at the first transfusion before hydroxyurea therapy and history of splenectomy are predictors of a good response tohydroxyurea 48. In a large study on 232 beta thalassemia intermedia patients in Iran a clinical and para clinical response to hydroxyurea was evaluated. Through this study optimal dose of hydroxyurea (8–15 mg/kg/d) was identified as well as the types of patients who would be most responsive to this treatment. It was proved that response to the drug was not determined solely by beta gene mutation, as previously believed, but that other clinical factors and genetic backgrounds other than the beta gene locus seem to be responsible for susceptibility to hydroxyurea response 49.
During 10 years of observation, hydroxyurea had no significant adverse effects in these patients such as malignancy, infertility, or bone marrow suppression when used at a dose of 8–15 mg/kg/d 50. However, further studies regarding possible long-term adverse effects of hydroxyurea are needed. Nevertheless, based on data from a large observational studies and small clinical trials, hydroxyurea can be considered in certain groups of patients. According to the Thalassemia International Federation, patients with beta thalassemia intermedia homozygous for the Xmnl polymorphism, patients with Lepore or δB-thalassemia, and patients for which a transfusion course is required but are alloimmunized could be considered for hydroxyurea therapy. Furthermore, patients with pulmonary hypertension, erythroid marrow hyperplasia and leg ulcers are also considered for hydroxyurea therapy.
Recombinant human erythropoietin (rHuEPO) is another erythropoiesis and HbF inducer that was shown to ameliorate the thalassemia clinical symptoms by increasing HbF cells and HbF induction. A dose related increase in erythropoiesis was seen in beta thalassemia intermedia patients by a dose of 500–1000 IU/kg × 3/week in several studies 51. A recent study also showed that recombinant human erythropoietin (rHuEPO) combined with hydroxyurea has a superior therapeutic effect in the clinical and hematological responses and improved quality of life in beta thalassemia intermedia patients in comparison with hydroxyurea therapy alone 52.
Splenectomy
Removal of the spleenmay be a useful treatment strategy in severe forms of beta thalassemia intermedia increasing the Hb levels by 1–2 g/dL and improving growth and development. Indications
for splenectomy in beta thalassemia intermedia are numerous, however, splenectomized patients are at greater risk of thrombosis, infection, and pulmonary hypertension 53, therefore, the decision to remove the spleen should be made with considerable caution. The Thalassemia International Federation guidelines on splenectomy indicate that splenectomy should be avoided in nontransfusion dependent thalassemia patients younger than 5 years of age. Furthermore, patients should undergo splenectomy in case of worsening anemia leading to poor growth and development, in cases of hypersplenism leading to worsening anemia, leucopenia, or thrombocytopenia. Also, splenomegaly accompanied by symptoms and massive splenomegaly with concern of splenic rupture should motivate physicians for the splenectomy procedure. Moreover, it is very important to keep in mind that abdominal sonography is mandatory before surgery because the gallbladder and accessory spleen should be inspected during splenectomy and removed in case of cholethiasis or accessory spleen.
Stem cell transplantation
Stem cell transplantation isn ot usually recommended in not transfused or irregularly transfused patients with beta thalassemia intermedia, although if these patients are transfusion dependent,
it may be considered.
Other treatment considerations
These include daily folic acid supplementation, antioxidant, anticaogulation inthe perioperative period, and adequate dietary or supplemental vitamin D and calcium for prevention of osteoporosis 3.
Beta thalassemia intermedia prognosis
Patients with thalassemias including beta thalassemia intermedia have some mental and physical complications that influence their health-related quality of life. These complications include the difficulties of living with a chronic disease, frequent physician and hospital visits, transfusion, chelation therapy, and family and social problems that are more prominent in developing countries. Because beta thalassemia intermedia is less severe than beta thalassemia major, some believe that health-related quality of life must be better. However, two studies of health-related quality of life in children and adults with beta thalassemia intermedia showed that transfusion-independent beta thalassemia intermedia patients actually have impaired health-related quality of life compared with beta thalassemia major patients of similar age and sex who receive regular transfusions 54, 55. These studies illustrate the importance of early diagnosis and appropriate management therapy of beta thalassemia intermedia, especially with regard to prompt recognition of the need for timely introduction of regular transfusions or chelation therapy and the provision of psychological support of patients and their families struggling with chronic disease.
In conclusion, although the clinical manifestations of beta thalassemia intermedia are usually milder that beta thalassemia major, the prognosis and the rate of complications are often worse 4. Right-sided heart failure due to long-standing pulmonary hypertension, thrombosis and brain ischemia, erythroid marrow hyperplasiain some vital regions like spinal cord, and iron overload are major and life-threatening complications of beta thalassemia intermedia. Close follow-up and monitoring of these patients for prevention of such complications and improved health-related quality of life are recommended.
Beta thalassemia
Beta thalassemia happens when the gene that controls the production of beta globin is defective. To date, more than 250 mutations that could cause beta thalassemia have been reported all over the world 56. Beta thalassemia can cause anemia ranging from mild to severe and is more common in people of Mediterranean, North African, the Middle Eastern, Indian, Central Asian, and Southeast Asian descent.
A child can only get beta thalassemia by inheriting it from his or her parents. Genes are “building blocks” that play an important role in determining physical traits and many other things about us.
Humans are made up of trillions of cells that form the structure of our bodies and carry out specialized jobs like taking nutrients from food and turning them into energy. Red blood cells, which contain hemoglobin, deliver oxygen to all parts of the body.
All cells have a nucleus at their center, which is kind of like the brain or “command post” of the cell. The nucleus directs the cell, telling it to grow, mature, divide, or die. The nucleus contains DNA (deoxyribonucleic acid), a long, spiral-shaped molecule that stores the genes that determine hair color, eye color, whether or not a person is right- or left-handed, and many more traits. DNA, along with genes and the information they contain, is passed down from parents to their children during reproduction.
Each cell has many DNA molecules, but because cells are very small and DNA molecules are long, the DNA is packaged very tightly in each cell. These packages of DNA are called chromosomes, and each cell has 46 of them. Each package is arranged into 23 pairs — with one of each pair coming from the mother and one from the father. When someone has beta thalassemia, there is a mutation in chromosome 11.
Beta globin is made on chromosome 11 (beta globin, along with alpha globin, is one of the proteins that makes up hemoglobin). So, if one or both of the genes that tells chromosome 11 to produce beta globin is altered, less beta globin is made. This affects hemoglobin and decreases the ability of red blood cells to transport oxygen around the body.
Beta thalassemia types
There are three types of beta thalassemia, depending upon whether one or two beta globin genes are mutated, and the severity of symptoms 57:
- Beta thalassemia minor, or beta thalassemia trait, happens when one of the beta globin genes is mutated. People with this condition typically have very mild symptoms and require no treatment, but they can pass thalassemia on to their children. Usually, they are mildly anemic and their red blood cells are smaller than normal.
- Beta thalassemia major (Cooley’s anemia) happens when both of the beta globin genes are mutated (changed) and the mutations are severe. This is the most severe form of beta thalassemia. Babies with beta thalassemia major often seem healthy immediately after birth but start to develop symptoms within the first 2 years of life. This condition causes severe symptoms with life-threatening anemia that requires regular blood transfusions.
- Beta thalassemia intermedia may also happen when both of the beta globin genes are mutated, but the mutations are less severe than those that typically cause beta thalassemia major. People with this condition usually have moderately severe anemia and sometimes require regular blood transfusions.
- Dominant beta thalassemia is an extremely rare form in which individuals who have one mutated HBB gene develop certain symptoms associated with beta thalassemia. Affected individuals may develop mild to moderate anemia, jaundice, and an abnormally enlarged spleen (splenomegaly).
The signs and symptoms of thalassemia major appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged spleen, liver, and heart, and their bones may be misshapen. Some adolescents with thalassemia major experience delayed puberty. Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body, resulting in liver, heart, and hormone problems.
Thalassemia intermedia is milder than thalassemia major. The signs and symptoms of thalassemia intermedia appear in early childhood or later in life. Affected individuals have mild to moderate anemia and may also have slow growth and bone abnormalities.
Beta thalassemia causes
Mutations in the HBB gene cause beta thalassemia. The HBB gene provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of hemoglobin. Hemoglobin consists of four protein subunits, typically two subunits of beta-globin and two subunits of another protein called alpha-globin.
Some mutations in the HBB gene prevent the production of any beta-globin. The absence of beta-globin is referred to as beta-zero (B0) thalassemia. Other HBB gene mutations allow some beta-globin to be produced but in reduced amounts. A reduced amount of beta-globin is called beta-plus (B+) thalassemia. Having either B0 or B+ thalassemia does not necessarily predict disease severity, however; people with both types have been diagnosed with thalassemia major and thalassemia intermedia.
A lack of beta-globin leads to a reduced amount of functional hemoglobin. Without sufficient hemoglobin, red blood cells do not develop normally, causing a shortage of mature red blood cells. The low number of mature red blood cells leads to anemia and other associated health problems in people with beta thalassemia.
Beta thalassemia inheritance pattern
Thalassemia major and thalassemia intermedia are inherited in an autosomal recessive pattern, which means both copies of the HBB gene in each cell have mutations (see Figure 1). The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene and are referred to as carriers, but they typically do not show signs and symptoms of the condition. When two carriers have children, each child has a 25% (1 in 4) chance to be affected, a 50% (1 in 2) chance to be a carrier like each parent, and a 25% (1 in 4) chance to be unaffected and not a carrier. Sometimes, however, people (carriers) with only one HBB gene mutation in each cell develop mild anemia. These mildly affected people are said to have ‘beta-thalassemia minor’ or ‘beta-thalassemia trait’ 58.
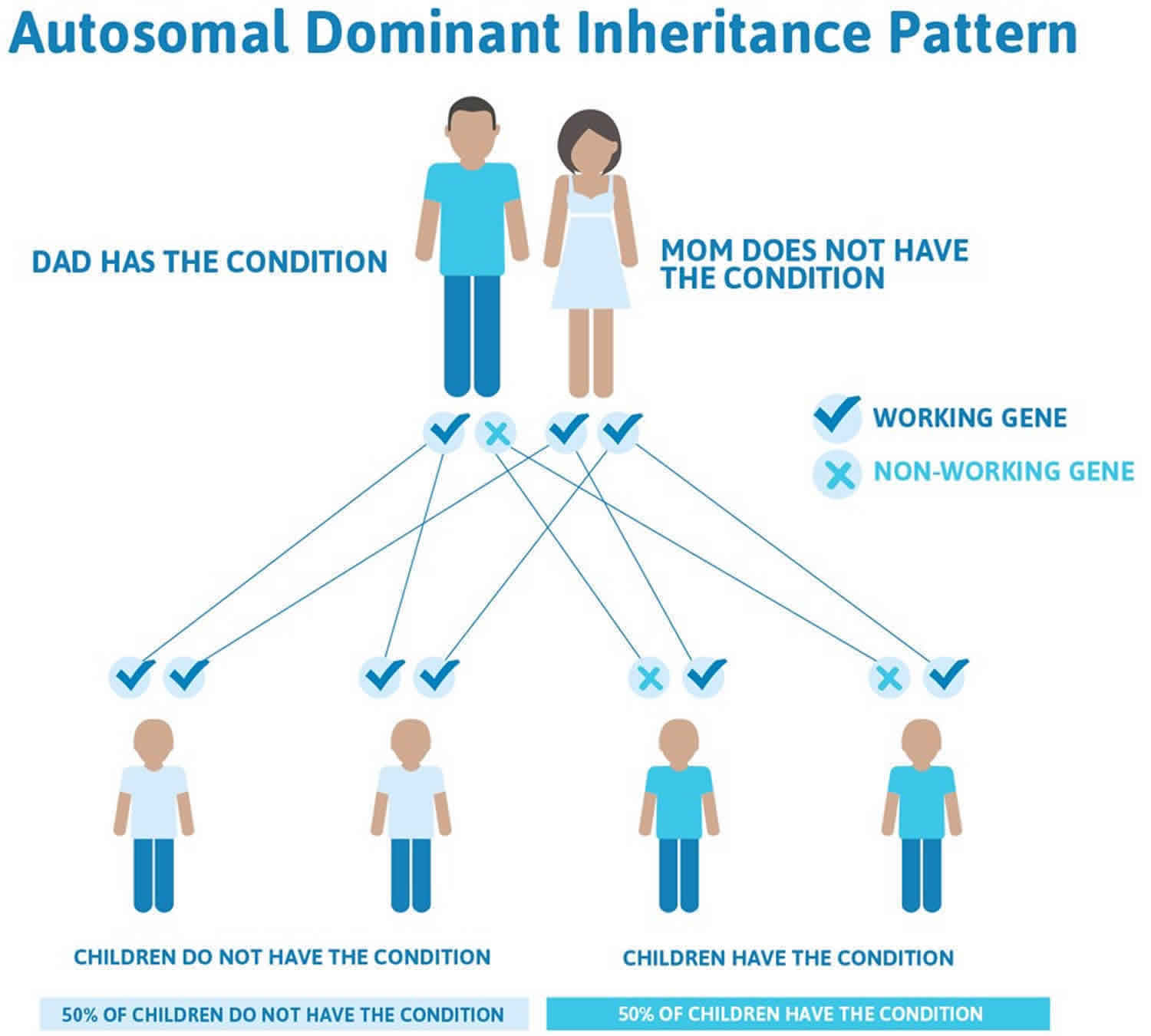
In a small percentage of families, the HBB gene mutation is inherited in an autosomal dominant manner (Figure 3). In these cases, one copy of the altered gene in each cell is sufficient to cause the signs and symptoms of beta thalassemia 58.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Beta thalassemia intermedia autosomal recessive inheritance pattern
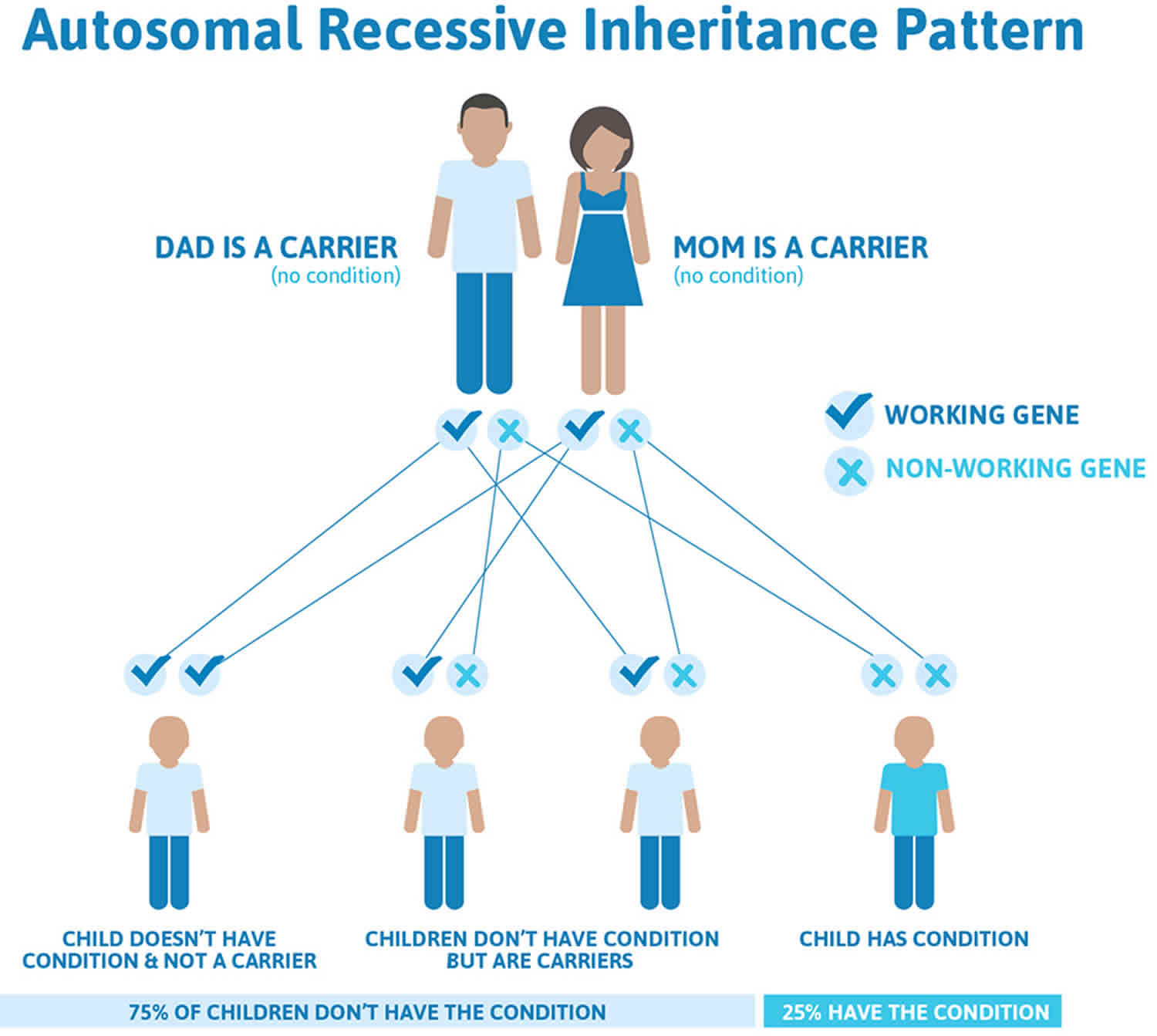
Figure 3. Beta thalassemia autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Beta thalassemia symptoms
The signs and symptoms of beta thalassemia vary depending on the type that a child has. Most children with beta thalassemia trait have no symptoms. Those with beta thalassemia major and intermedia may not show any symptoms at birth, but usually develop them in the first 2 years of life.
Some of the more common symptoms of beta thalassemia include:
- fatigue, weakness, or shortness of breath
- a pale appearance or a yellow color to the skin (jaundice)
- irritability
- deformities of the facial bones
- slow growth
- a swollen abdomen
- dark urine
Babies who begin to show symptoms of beta thalassemia after a few healthy months may fail to grow normally (failure to thrive); have trouble feeding; and have episodes of fever, diarrhea, and other intestinal problems.
Beta thalassemia complications
Beta thalassemia major and intermedia can lead to serious complications, especially if untreated. Complications of beta thalassemia major include:
- Excess iron. Kids who have beta thalassemia can end up with too much iron in their bodies, either from the disease itself or from getting repeated blood transfusions. Excess iron can cause damage to the heart, liver, and endocrine system.
- Bone deformities and broken bones. Beta thalassemia can cause bone marrow to expand, making bones wider, thinner, and more brittle. This makes bones more likely to break and can lead to abnormal bone structure, particularly in the bones of the face and skull.
- Enlarged spleen. The spleen helps fight off infections and filters out unwanted materials, such as dead or damaged blood cells, from the body. Beta thalassemia can cause red blood cells to die off at a faster rate, making the spleen work harder, which makes it grow larger. A large spleen can make anemia worse and may need to be removed if it gets too big.
- Infections. Children with beta thalassemia have a higher risk of infection, especially if they’ve had their spleens removed.
- Slower growth rates. The anemia resulting from beta thalassemia can cause children to grow more slowly and also can lead to delayed puberty.
- Extramedullary hematopoiesis
- Medical complications from long-term transfusional therapy – Iron overload and transfusion-associated infections (eg, hepatitis); iron overload cardiomyopathy accounts for the majority of deaths in thalassemia patients 59.
- Increased risk for infections resulting from asplenia (eg, encapsulated organisms such as pneumococcus) or from iron overload (eg, Yersinia species)
- Cholelithiasis (eg, bilirubin stones)
Beta thalassemia diagnosis
In most cases, beta thalassemia is diagnosed before a child’s second birthday. Children with beta thalassemia major may have a swollen abdomen or symptoms of anemia or failure to thrive.
If the doctor suspects beta thalassemia, he or she will take a blood sample for testing. Blood tests can reveal red blood cells that are pale, varied in shape and size, and smaller than normal. They also can detect low red blood cell counts and cells with an uneven distribution of hemoglobin, which causes them to look like a bull’s-eye when seen through a microscope.
Blood tests also can measure the amount of iron in the blood. Usually the diagnosis is confirmed by a blood test called a hemoglobin electrophoresis and/or a DNA test for abnormal hemoglobin genes.
Molecular genetic testing can confirm a beta thalassemia diagnosis. Molecular genetic testing can detect mutations in the HBB gene known to cause the disorder, but is available only as a diagnostic service at specialized laboratories. Molecular genetic testing is not necessary to make a diagnosis of beta thalassemia and is generally used to identify at-risk, asymptomatic relatives, to aid prenatal diagnosis, and to attempt to predict the progression or severity of the disease in specific cases.
If both parents are carriers of the beta thalassemia disorder, doctors can conduct tests on a fetus before birth. This is done through either:
- chorionic vilius sampling, which takes place about 11 weeks into pregnancy and involves removing a tiny piece of the placenta for testing
- amniocentesis, which is usually done about 16 weeks into the pregnancy and involves removing a sample of the fluid that surrounds the fetus
If one parent carries a beta thalassemia gene and the other carries a different gene that also affects beta globin, such as a sickle gene, their child could have a significant blood disorder (such as a form of sickle cell disease called sickle-beta thalassemia). Therefore, people who carry beta thalassemia genes should seek genetic counseling if they’re considering having children so they can understand the risks.
Beta thalassemia treatment
Individuals with beta thalassemia major and intermedia will benefit from referral to a thalassemia treatment center. These specialized centers provide comprehensive care for individuals with beta thalassemia including the development of specific treatment plans, monitoring and follow up of affected individuals, and state-of-the-art medical care. Treatment at such a center ensures that individuals and their family members will be cared for by a professional healthcare team (physicians, nurses, physical therapists, social workers and genetic counselors) experienced in the treatment of individuals with beta thalassemia. Genetic counseling is recommended for affected individuals and their families. Psychosocial support for the entire family is essential as well.
Specific therapeutic procedures and interventions may vary, depending upon numerous factors, such as the specific type of beta thalassemia; the progression of the disease; the presence or absence of certain symptoms; severity of the disease upon diagnosis; an individual’s age and general health; and/or other elements. Decisions concerning the use of a particular drug regimen and/or other treatments should be made by physicians and other members of the health care team in consultation with the patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
The amount of treatment that beta thalassemia requires depends on how severe the symptoms are. For most children with beta thalassemia trait, whose only symptom may be mild anemia from time to time, no medical treatment will be necessary. It is important that individuals with beta thalassemia minor be correctly diagnosed, however, in order to avoid unnecessary treatments for similarly-appearing conditions such as iron deficiency anemia.
However, the blood counts in beta thalassemia trait look a lot like the blood counts in iron deficiency anemia, which is a very common disorder. It’s important for doctors to know when children have beta thalassemia trait so that they do not treat them with iron if it’s not needed.
Doctors also might recommend a folic acid supplement for kids with moderate cases of anemia to help boost production of new red blood cells. Supplementation with folic acid, a B vitamin, boosts the production of red blood cells in certain individuals.
Some children with moderate anemia may require an occasional blood transfusion, particularly after surgery. Those with severe cases of beta thalassemia major, on the other hand, may require regular blood transfusions their entire lives to keep them healthy. During blood transfusions, they’re given blood from donors with matching blood types. Over time, this can cause a build-up of iron in the body, so kids who receive frequent blood transfusions may have to take medications to remove excess iron from their bodies.
Some individuals may be treated by the surgical removal of the spleen (splenectomy). An abnormally enlarged spleen (splenomegaly) can cause severe pain and contribute to anemia. Splenomegaly can cause low levels of the blood cells (platelets) that allow the blood to clot. An enlarged spleen in individuals with beta thalassemia may occur due to increased destruction of red blood cells, the formation of blood cells outside of the bone marrow (extramedullary hematopoiesis), repeated blood transfusions, or iron overload. If other forms of therapy fail, removal of the spleen may be considered. Splenectomy has led to improvement in certain symptoms associated with beta thalassemia. However, this surgical procedure carries risks, which are weighed against benefits in each individual case. If a splenectomy is required, one month before the surgery pneumococcal conjugate vaccine should be given. In addition, antibiotic prophylaxis, usually penicillin 250 mg twice a day, is given the first two years after surgery and for children younger than 16 years. Because of advances in the treatment of beta thalassemia in the past several years, splenectomy is rarely necessary as a treatment for affected individuals.
Individuals with beta thalassemia major and intermedia may develop iron overload, which occurs because of two reasons. First, blood transfusions cause the accumulation of excess iron in the body. Second, beta thalassemia can cause increased absorption of dietary iron by the gastrointestinal tract. The body has no normal way to remove excess iron. In individuals who receive regular blood transfusions, iron overload primarily occurs because of treatment. Iron overload causes a variety of symptoms affecting various body organ systems. Iron overload is treated by medications that remove excess iron from the body such as deferoxamine. Deferoxamine is an iron chelator, a drug that binds to iron in the body allowing it to be dissolved in water and excreted from the body through the kidneys. Other oral iron chelators, such as deferiprone and deferasirox, have also been used to lower iron levels.
Treatment of additional complications of beta thalassemia or iron overload is symptomatic and supportive. Special attention is recommended for the early diagnosis and prompt treatment of heart (cardiac) disease potentially associated with iron overload. Cardiac disease is the main life-threatening complication in individuals with beta thalassemia.
Research into treating beta thalassemia with experimental gene therapies is ongoing. But for now, it can only be cured by a procedure called a bone marrow transplant (also called a stem cell transplant). Bone marrow, which is found inside bones, produces blood cells. In a bone marrow transplant, children are first given high doses of radiation or drugs to destroy the defective bone marrow. The bone marrow is then replaced with cells from a compatible donor, usually a healthy sibling or other relative. Bone marrow transplants carry many risks, so they usually are done only in the most severe cases of thalassemia.
If your child has beta thalassemia, support groups are available to help your family cope with the obstacles presented by the disease.
Alpha thalassemia
Normally, each person has four genes for alpha globin. Alpha thalassemia happens when one or more of the genes that control the making of alpha globins is absent or defective. It can cause anemia ranging from mild to severe and is most commonly found in people of African, Middle Eastern, Chinese, Southeast Asian, and, occasionally, Mediterranean descent.
Some children with alpha thalassemia have no symptoms and require no treatment. Others with more severe cases need regular blood transfusions to treat anemia and other symptoms.
A child can only get alpha thalassemia by inheriting it from his or her parents. Genes are “building blocks” that play an important role in determining physical traits and many other things about us.
Humans are made up of trillions of cells that form the structure of our bodies and carry out specialized jobs like taking nutrients from food and turning them into energy. Red blood cells, which contain hemoglobin, deliver oxygen to all parts of the body.
All cells have a nucleus at their center, which is kind of like the brain or “command post” of the cell. The nucleus directs the cell, telling it to grow, mature, divide, or die. The nucleus contains DNA (deoxyribonucleic acid), a long, spiral-shaped molecule that stores the genes that determine hair color, eye color, whether or not a person is right- or left-handed, and many more traits. DNA, along with genes and the information they contain, is passed down from parents to their children during reproduction.
Each cell has many DNA molecules, but because cells are very small and DNA molecules are long, the DNA is packaged very tightly in each cell. These packages of DNA are called chromosomes, and each cell has 46 of them. Each package is arranged into 23 pairs — with one of each pair coming from the mother and one from the father. When a child has alpha thalassemia, there is a change in chromosome 16.
Alpha globin is made on chromosome 16. So, if any gene that tells chromosome 16 to produce alpha globin is missing or mutated, less alpha globin is made. This affects hemoglobin and decreases the ability of red blood cells to transport oxygen around the body.
Alpha thalassemia causes
Alpha thalassemia typically results from deletions involving the HBA1 and HBA2 genes. Both of these genes provide instructions for making a protein called alpha-globin, which is a component (subunit) of hemoglobin.
People have two copies of the HBA1 gene and two copies of the HBA2 gene in each cell. Each copy is called an allele. For each gene, one allele is inherited from a person’s father, and the other is inherited from a person’s mother. As a result, there are four alleles that produce alpha-globin. The different types of alpha thalassemia result from the loss of some or all of these alleles.
Hb Bart syndrome, the most severe form of alpha thalassemia, results from the loss of all four alpha-globin alleles. HbH disease is caused by a loss of three of the four alpha-globin alleles. In these two conditions, a shortage of alpha-globin prevents cells from making normal hemoglobin. Instead, cells produce abnormal forms of hemoglobin called hemoglobin Bart (Hb Bart) or hemoglobin H (HbH). These abnormal hemoglobin molecules cannot effectively carry oxygen to the body’s tissues. The substitution of Hb Bart or HbH for normal hemoglobin causes anemia and the other serious health problems associated with alpha thalassemia.
Two additional variants of alpha thalassemia are related to a reduced amount of alpha-globin. Because cells still produce some normal hemoglobin, these variants tend to cause few or no health problems. A loss of two of the four alpha-globin alleles results in alpha thalassemia trait. People with alpha thalassemia trait may have unusually small, pale red blood cells and mild anemia. A loss of one alpha-globin allele is found in alpha thalassemia silent carriers. These individuals typically have no thalassemia-related signs or symptoms.
Alpha thalassemia inheritance pattern
The inheritance of alpha thalassemia is complex. Each person inherits two alpha-globin alleles from each parent. If both parents are missing at least one alpha-globin allele, their children are at risk of having Hb Bart syndrome, HbH disease, or alpha thalassemia trait. The precise risk depends on how many alleles are missing and which combination of the HBA1 and HBA2 genes is affected.
Alpha thalassemia types
Alpha globin is made by four genes and one or more can be mutated or missing, so there are four kinds of alpha thalassemia:
- One missing or abnormal gene makes a child a silent alpha thalassemia carrier. Silent alpha thalassemia carriers have no signs or symptoms of the disease, but are able to pass thalassemia on to their children.
- Two missing or mutated genes is a condition called alpha thalassemia minor or having alpha thalassemia trait. Children with this condition may have red blood cells that are smaller than normal (microcytosis) and sometimes very slight anemia.People with alpha thalassemia minor usually don’t have any symptoms at all, but can pass thalassemia on to their children. The two abnormal genes can be on the same chromosome (called the cis position) or one on each chromosome (called the trans position). If two genes on the same chromosome are affected, the person can pass along a two-gene defect to his or her child. This situation is much more common in people of Asian descent.
- Three missing or mutated genes is called hemoglobin H disease. Signs and symptoms will be moderate to severe.
- Four missing or mutated genes is a condition known as alpha thalassemia major or hydrops fetalis. This almost always leads to a fetus dying before delivery or a newborn baby dying shortly after birth. However if this disease is suspected because of a history in the family, it can be diagnosed prenatally. Sometimes, if treatment is initiated before the baby is even born, the baby can survive.
Alpha thalassemia symptoms
The signs and symptoms of alpha thalassemia vary depending on the type that a child has and how severe it is. Children with alpha thalassemia trait and those who are silent carriers have no symptoms at all.
Some of the more common symptoms of alpha thalassemia include:
- fatigue, weakness, or shortness of breath
- a pale appearance or a yellow color to the skin (jaundice)
- irritability
- deformities of the facial bones
- slow growth
- a swollen abdomen
- dark urine
Alpha thalassemia complications
In addition to anemia and hydrops fetalis, severe cases of alpha thalassemia and hemoglobin H disease can lead to serious complications, especially if untreated. Complications of alpha thalassemia include:
- Excess iron. When children have alpha thalassemia, they can end up with too much iron in their bodies, either from the disease itself or from getting repeated blood transfusions. Excess iron can cause damage to the heart, liver, and endocrine system.
- Bone deformities and broken bones. Alpha thalassemia can cause bone marrow to expand, making bones wider, thinner, and more brittle. This makes bones more likely to break and can lead to abnormal bone structure, particularly in the bones of the face and skull.
- Enlarged spleen. The spleen helps fight off infections and filters out unwanted materials, such as dead or damaged blood cells, from the body. Alpha thalassemia can cause red blood cells to die off at a faster rate, making the spleen work harder, which makes it grow larger. A large spleen can make anemia worse and may need to be removed if it gets too big.
- Infections. Children with alpha thalassemia have an increased risk of infection, especially if they’ve had their spleens removed.
- Slower growth rates. The anemia resulting from alpha thalassemia can cause children to grow more slowly and also can lead to delayed puberty.
Alpha thalassemia diagnosis
In most cases, alpha thalassemia is diagnosed before a child’s second birthday or through newborn screening, a blood test given when the child is first born. Children with alpha thalassemia major may have a swollen abdomen or symptoms of anemia or failure to thrive.
If the doctor suspects alpha thalassemia, he or she will take a blood sample for testing. Blood tests can reveal red blood cells that are pale, varied in shape and size, or smaller than normal. They also can detect low red blood cell counts and cells with an uneven distribution of hemoglobin, which causes them to look like a bull’s-eye when seen through a microscope.
Blood tests also can measure the amount of iron in the blood, evaluate hemoglobin, and test a child’s DNA for abnormal hemoglobin genes.
If both parents are carriers of the alpha thalassemia disorder, doctors can conduct tests on a fetus before birth. This is done through either:
- chorionic vilius sampling, which takes place about 11 weeks into pregnancy and involves removing a tiny piece of the placenta for testing
- amniocentesis, which is usually done about 16 weeks into the pregnancy and involves removing a sample of the fluid that surrounds the fetus
Alpha thalassemia treatment
The amount of treatment that alpha thalassemia requires depends on how severe the symptoms are. For those with alpha thalassemia trait or silent carriers with only mild anemia from time to time, no medical treatment is necessary.
However, the blood counts in alpha thalassemia trait look a lot like the blood counts in iron deficiency anemia, which is a very common disorder. It’s important for doctors to know when children have alpha thalassemia trait so that they do not treat them with iron if it’s not needed.
Doctors also might recommend a folic acid supplement for kids with hemoglobin H disease to help the body make new red blood cells. In addition, these kids may require an occasional blood transfusion, particularly after surgery.
Less commonly, children with severe cases of hemoglobin H disease may require regular blood transfusions their entire lives to keep them healthy. During blood transfusions, they’re given blood from donors with matching blood types. Over time, this can cause a build-up of iron in the body, so kids who receive frequent blood transfusions may have to take medications to remove excess iron from their bodies.
Currently, the only cure for thalassemia is a procedure called a bone marrow transplant (also called a stem cell transplant). Bone marrow, which is found inside bones, produces blood cells. In a bone marrow transplant, a person is first given high doses of radiation or drugs to destroy the defective bone marrow. The bone marrow is then replaced with cells from a compatible donor, usually a healthy sibling or other relative. Bone marrow transplants carry many risks, so they usually are done only in the most severe cases of thalassemia.
Because living with alpha thalassemia can be challenging, people who are carriers of alpha thalassemia trait may want to seek genetic counseling if they’re considering having children.
If your child has alpha thalassemia, support groups are available to help your family cope with the obstacles presented by the disease.
References- Taher A, Vichinsky E, Musallam Ket al., authors; Weatherall D, editor. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT) [Internet]. Nicosia, Cyprus: Thalassaemia International Federation; 2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK190453
- Ansari S, Rashid N, Hanifa A, et al. Laboratory diagnosis for thalassemia intermedia: Are we there yet?. J Clin Lab Anal. 2019;33(1):e22647. doi:10.1002/jcla.22647 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6430353
- Taher A,Musallam K, Cappellini MD,Weatherall DJ. Optimalmanagement of β thalassaemia intermedia. Br J Haematol. 2011;152:512–523.
- Karimi, Mehran & Cohan, Nader & Sanctis, Vincenzo & Mallat, Naji & Taher, Ali. (2014). Guidelines for Diagnosis and Management of Beta-Thalassemia Intermedia. Pediatric hematology and oncology. 31. 583-596. 10.3109/08880018.2014.937884.
- Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: Revisited. Blood Cells Mol Dis. 2006;37:12–20.
- Galanello R, Cao A. Relationship between genotype and phenotype. Thalassemia intermedia. Ann NY Acad Sci 1998;850:325-33.
- Camaschella C, Mazza U, Roetto A, et al. Genetic interactions in thalassemia intermedia: analysis of β-mutations, alpha-genotype, gamma-promoters, andβ-LCR hypersensitive sites 2 and 4 in Italian patients. Am J Hematol 1995;48:82-7.
- Xie J, Zhou Y, Xiao Q, Long R, Li L, Li L. Rare double heterozygosity for poly A(A>G) and CD17(A>T) of beta thalassemia intermedia in a Chinese family. Hematol Rep. 2019;11(3):7911. Published 2019 Sep 18. doi:10.4081/hr.2019.7911 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6761475
- Thein SL. Pathophysiology of β thalassemia—A guide to molecular therapies. Hematology Am Soc Hematol Educ Program. 2005;2005(1):31‐37.
- El Rassi F, Cappellini MD, Inati A, Taher A. Beta-thalassemia intermedia: An overview. Pediatric Annals. 2008;37(5):322–328.
- Karimi, Mehran & Cohan, Nader & Sanctis, Vincenzo & Mallat, Naji & Taher, Ali. (2014). Guidelines for Diagnosis and Management of Beta-Thalassemia Intermedia. Pediatric hematology and oncology. 31. 583-596. 10.3109/08880018.2014.937884.
- Cao A, Galanello R. Beta‐thalassemia. Genet Med. 2010;12(2):61.
- Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis 2006;37:12-20.
- Taher AT, Musallam KM, Cappellini MD. Thalassaemia intermedia: an update. Mediterr J Hematol Infect Dis. 2009;1(1):e2009004
- Kaddah N, Salama K, Kaddah AM, Attia R. Epidemiological study among thalassemia intermedia pediatric patients. Med J Cairo Univ. 2010;78(2):651‐655.
- Taher A, Cappellini MD, Ismaeel H. Thalassemia: the facts and the controversies. Pediatr Nurs. 2006;29(6):447‐451.
- Qatanani M, Taher A, Koussa S, et al. β‐Thalassaemiaintermedia in Lebanon. Eur J Haematol. 2000;64(4):237‐244.
- Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37(1):12‐20.
- Taher AT, Radwan A, Viprakasit V. When to consider transfusion therapy for patients with non‐transfusion‐dependent thalassaemia. Vox Sang. 2015;108(1):1.
- Taher A, Isma’eel H, Mehio G, et al. Prevalence of thromboembolic events among 8,860 patients with thalas- saemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96:488–491.
- Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892.
- Taher AT, Musallam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8(10):2152–2158.
- Musallam KM, Taher AT, Karimi M, Rachmilewitz EA. Cerebral infarction in β-thalassemia intermedia: breaking the silence. Thromb Res. 2012;130(5):695–702.
- Manfr`e L, Giarratano E, Maggio A, et al. MR imaging of the brain: findings in asymptomatic patients with thalassemia intermedia and sickle cell-thalassemia disease. AJR Am J Roentgenol. 1999;173(6):1477–1480.
- Taher AT, Musallam KM, Nasreddine W, et al. Asymptomatic brain magnetic resonance imaging abnormalities in splenectomized adults with thalassemia intermedia. J Thromb Haemost. 2010;8(1):54–59.
- Karimi M, Bagheri H, Rastgu F, Rachmilewitz E. Magnetic resonance imaging to determine the incidence of brain ischaemia in patients with β-thalassaemia intermedia. Thromb Haemost. 2010;103:989–993.
- Karimi M, Haghpanah S, Bagheri MH, et al. Frequency and distribution of asymptomatic brain lesions in patients with β-thalassemia intermedia. Ann Hematol. 2012;91(12):1833–1838.
- Karimi M, Musallam KM, Cappellini MD, et al. Risk factors for pulmonary hypertension in patients with β thalassemia intermedia. Eur J Intern Med. 2011;22(6):607–610.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359:2254–2265.
- Aessopos A, Farmakis D, Tsironi M, et al. Endothelial function and arterial stiffness in sicklethalassemia patients. Atherosclerosis. 2007;191:427–432.
- Amoozgar H, Farhani N, Karimi M. Risk factors for pulmonary hypertension in patients with thalassemia intermedia. Eur J Haematol. 2011;85:549–551.
- De Sanctis V, Tangerini A, TestaMR, et al. Final height and endocrine function in thalassemia intermedia. J Pediatr Endocrinol Metab. 1998;11(Suppl 3):965–971.
- Harmatz P, Jonas MM, Kwiatkowski JL, et al. Safety and efficacy of pegylated interferon a-2a and ribavirin for the treatment of hepatitis C in patients with thalassemia. Haematologica. 2008;93:1247–1251.
- Restivo Pantalone G, Renda D, Valenza F, et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: clinical characteristics and outcome in a long term single centre experience. Br J Haematol. 2010;(150):245–247.
- Matta BN, Abbas O, Maakaron JE, et al. Leg ulcers in patients with β-thalassaemia intermedia: a single centre’s experience. J Eur Acad Dermatol Venereol. 2013; [Epub ahead of print].
- Derakhshan A, Karimi M, Moghaddam AG. Comparative evaluation of renal finding in betathalassemiamajor and intermedia. Saudi J Kidney Dis Transpl. 2008;19(2):206–209.
- Rachid H, Hani M, Ali TT. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J. 2010;19(6):871–878.
- Karimi M, Cohan N, Bagheri MH, et al. A lump on the head. Lancet. 2008;372:1436.
- Karimi M, Cohan N, Pishdad P. Hydroxyurea as a first-line treatment of extramedullary hematopoiesis in patients with beta thalassemia: Four case reports. Hematology. 2014; [Epub ahead of print].
- Karimi M, Nikrooz P, Kashef S, et al. RBC alloimmunization in blood transfusion-dependent betathalassemia patients in southern Iran. Int J Lab Hematol. 2007;29(5):321–326.
- Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in nontransfusion- dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92(11):1485–1493.
- Musallam KM, Cappellini MD, Wood JC, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica 2011; 96(11): 1605–1612.
- Voskaridou E, Plata E, Douskou M, et al. Treatment with deferasirox (Exjade) effectively decreases iron burden in patients with thalassaemia intermedia: results of a pilot study. Brit J Haematol. 2010;148:332–334.
- Haghpanah S, Zarei T, Zahedi Z, Karimi M. Compliance and satisfaction with deferasirox (Exjade R) compared with deferoxamine in patients with transfusion-dependent beta-thalassemia. Hematology. 2014;19:187–189.
- Derakhshan A, Karimi M, Moghaddam AG. Comparative evaluation of renal finding in beta thalassemia major and intermedia. Saudi J Kidney Dis Transpl. 2008;19(2):206–209.
- Karimi M, Darzi H, Yavarian M. Hematologic and clinical responses of thalassemia intermedia patients to hydroxyurea during 6 years of therapy in Iran. J Ped Hematol Oncol. 2005;27:380–385.
- Karimi M,Mohammadi F, Behmanesh F, et al. Effect of combination therapy of hydroxyurea with l-carnitine and magnesium chloride on hematologic parameters and cardiac function of patients with beta-thalassemia intermedia. Eur J Haematol. 2010; 84: 52–58.
- Karimi M, Cohan N, Moosavizadeh K, et al. Adverse effects of hydroxyurea in β-thalassemia intermedia patients: 10 years’ experience. Ped Hematol Oncol. 2010;27:205–211.
- Karimi M, Haghpanah S, Farhadi A, Yavarian M. Genotype-phenotype relationship of patients with β-thalassemia taking hydroxyurea: a 13-year experience in Iran. Int J Hematol. 2012;95(1):51–56.
- Karimi M, ZekavatOR,Haghpanah S, et al. Comparative study of hypogonadism in beta-thalassemia intermedia patients with and without hydroxyurea. Hematology. 2012;17(2):122–124.
- Rachmilewitz EA, Aker M.The role of recombinant human erythropoietin in the treatment of thalassemia. Ann N Y Acad Sci. 1998;850:129–138.
- Elalfy MS, Adly AA, Ismail EA, et al.Therapeutic superiority and safety of combined hydroxyureawith recombinant human erythropoietin over hydroxyurea in young β-thalassemia intermedia patients. Eur J Haematol. 2013;91:522–533.
- Taher AT,Musallam KM, Karimi M, Cappellini MD. Contemporary approaches to treatment of betathalassemia intermedia. Blood Rev. 2012; 26(Suppl 1):S24–S27.
- Musallam KM, Khoury B, Abi-Habib R, et al.Health-related quality of life in adults with transfusion independent thalassaemia intermedia compared to regularly transfused thalassaemia major: new insights. Eur J Hematol. 2011;87:73–79.
- Pakbaz Z, Treadwell M, Yamashita R, et al. Quality of life in patients with thalassemia intermedia compared to thalassemiamajor. Ann N Y Acad Sci. 2005;1054:457–461.
- Xie J, Zhou Y, Xiao Q, Long R, Li L, Li L. Rare double heterozygosity for poly A(A〉 G) and CD17(A〉 T) of beta thalassemia intermedia in a Chinese family. Hematol Rep. 2019;11(3):7911. Published 2019 Sep 18. doi:10.4081/hr.2019.7911 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6761475
- Origa R. Beta-Thalassemia. 2000 Sep 28 [Updated 2018 Jan 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1426
- Beta thalassemia. https://ghr.nlm.nih.gov/condition/beta-thalassemia
- Siri-Angkul N, Chattipakorn SC, Chattipakorn N. Diagnosis and treatment of cardiac iron overload in transfusion-dependent thalassemia patients. Expert Rev Hematol. 2018 Jun. 11 (6):471-479.





{kind=link}